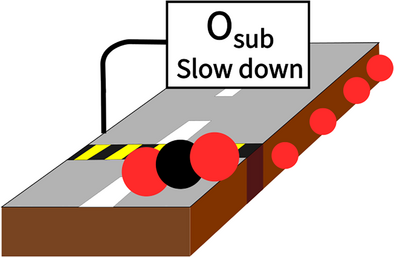
Controlling Heterogeneous Catalysis With Subsurface Oxygen
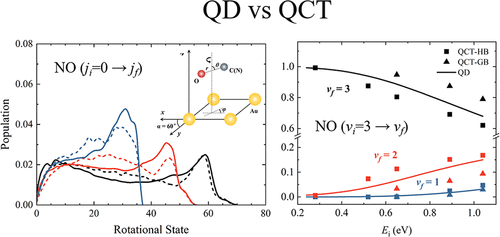
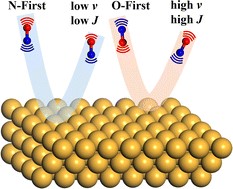
Six-Dimensional State-to-State Scattering of CO and NO from Au(111): A Comparison of Quantum and Quasi-Classical Dynamics
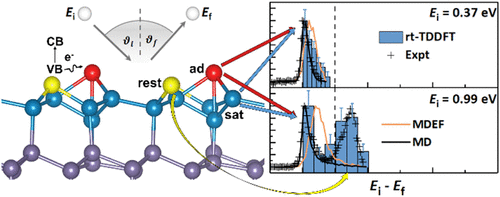
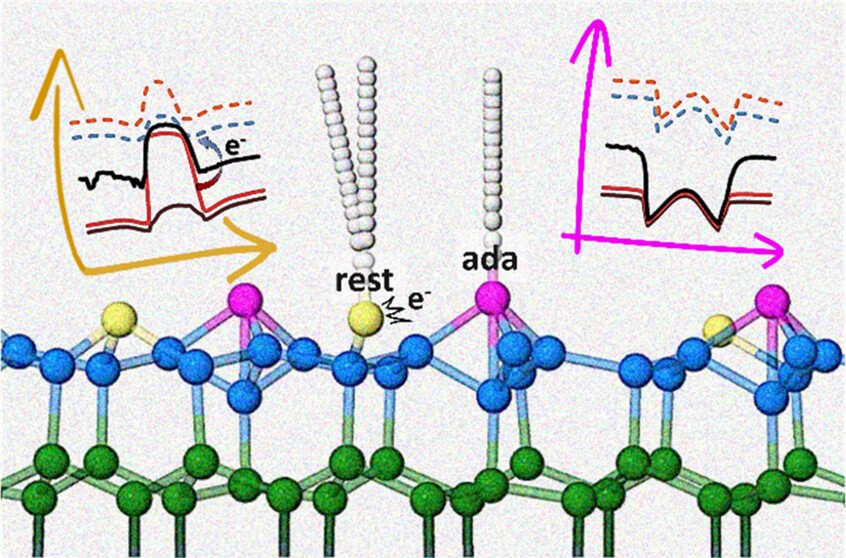
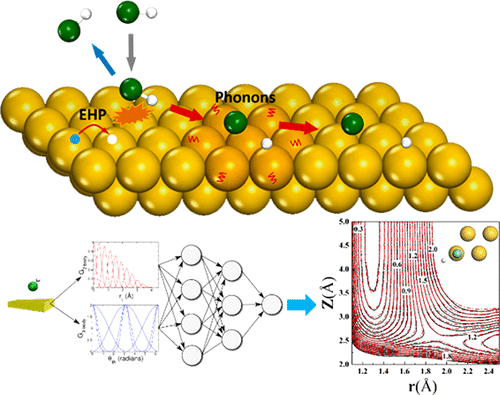
Real-Time Coupled Electron-Nuclear Dynamics of Chemical Bond Formation on a Semiconductor Surface
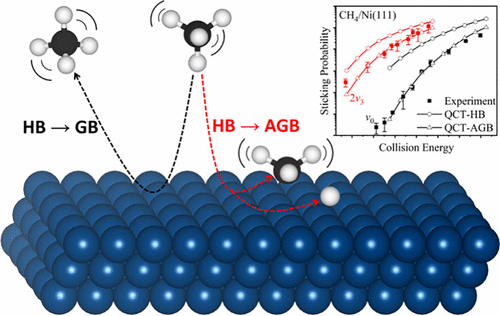
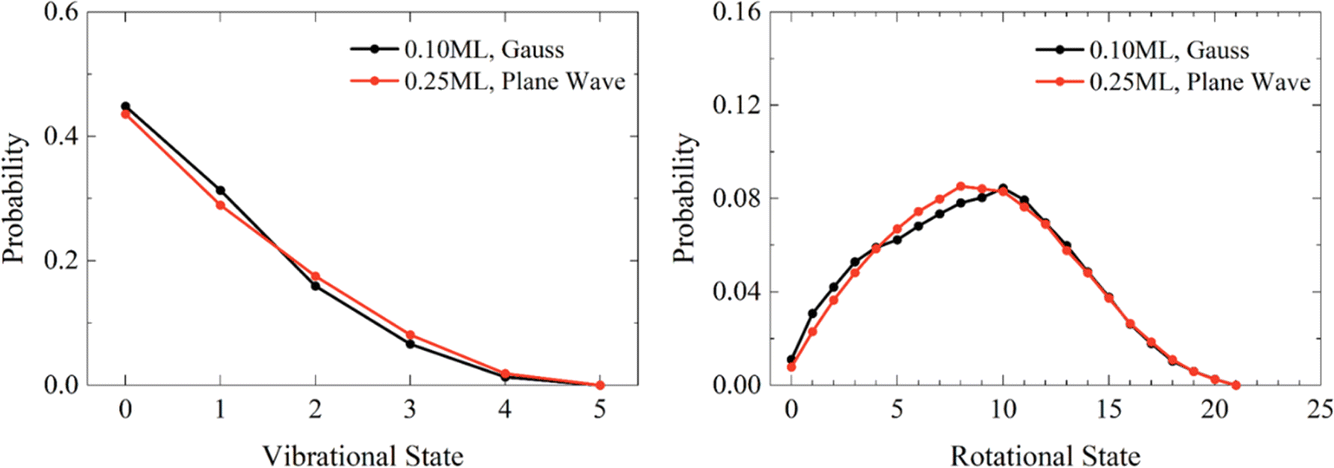
Quasi-Classical Trajectory with Adsorbate Gaussian Binning: Quantum-State-Resolved Prediction of Dissociative Sticking Probability Made Simple
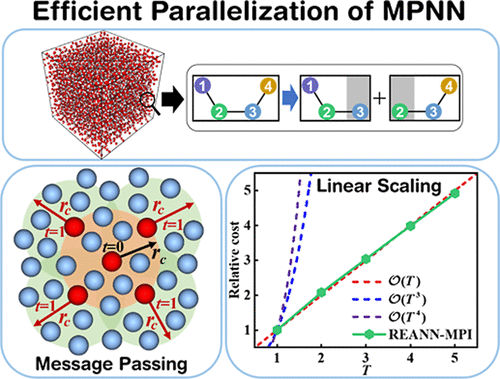
Efficient Parallelization of Message Passing Neural Network Potentials for Large-Scale Molecular Dynamics
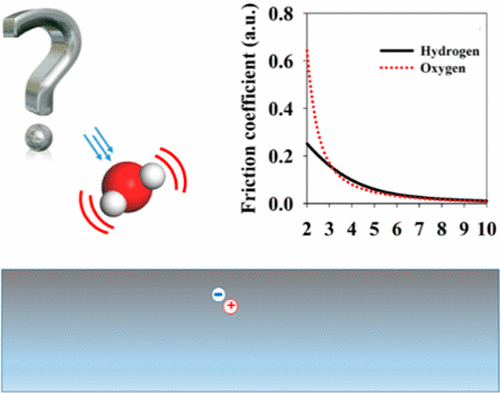
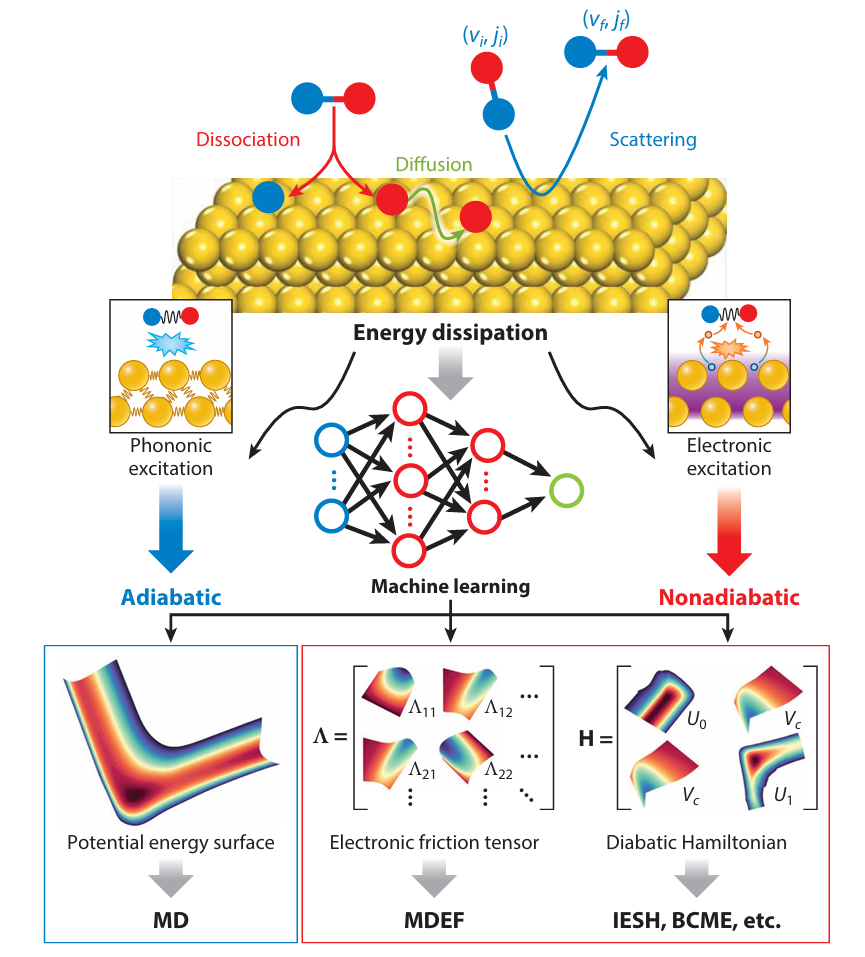
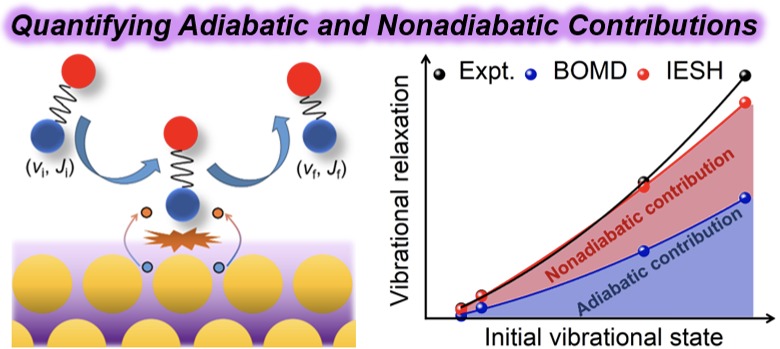
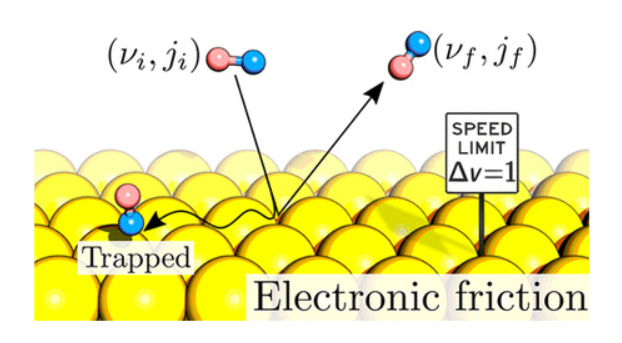
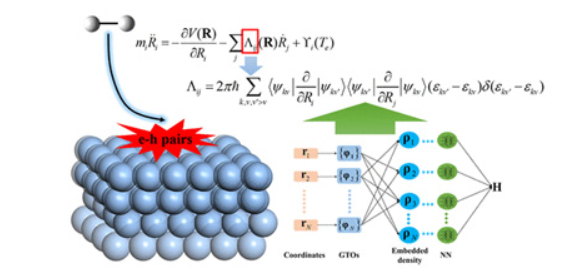
Dynamics of Surface Processes: Impact of Adiabatic and Nonadiabatic Energy Dissipation
Recent progress on embedded atom neural network approaches
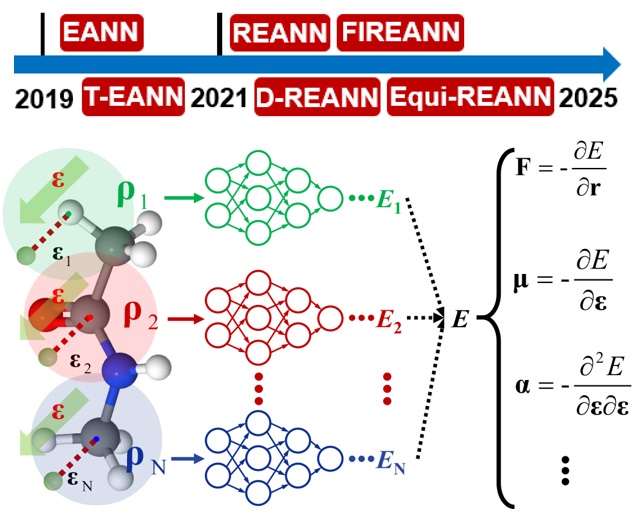
REANN 2.0: An Efficient Package of Neural Network Potentials for Multi-Element Systems

First-principles full-dimensional modelling of vibrational energy transfer of molecule scattering from metal surfaces
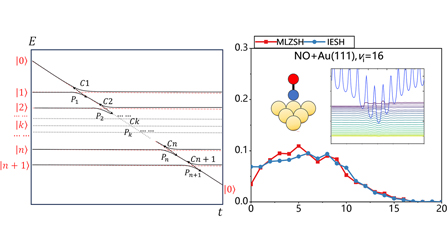
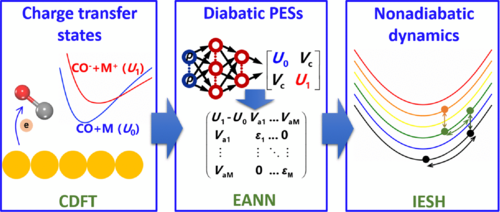
A multistate Landau–Zener surface hopping model for nonadiabatic dynamics of molecular scattering from metal surfaces
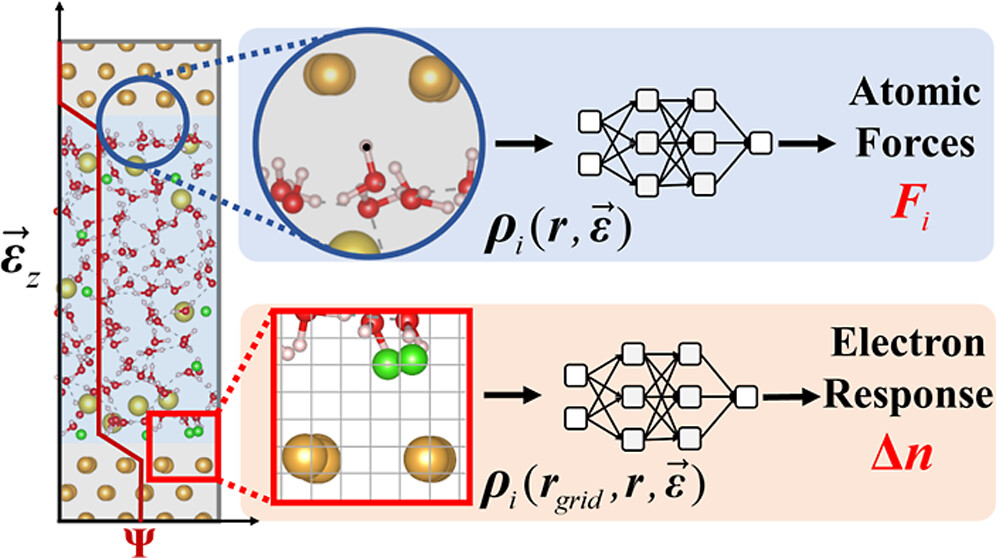
Machine Learning Accelerated Finite-Field Simulations for Electrochemical Interfaces
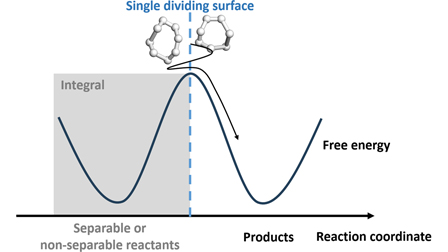
Unified ring polymer molecular dynamics rate calculations for reactions with separable and non-separable reactants
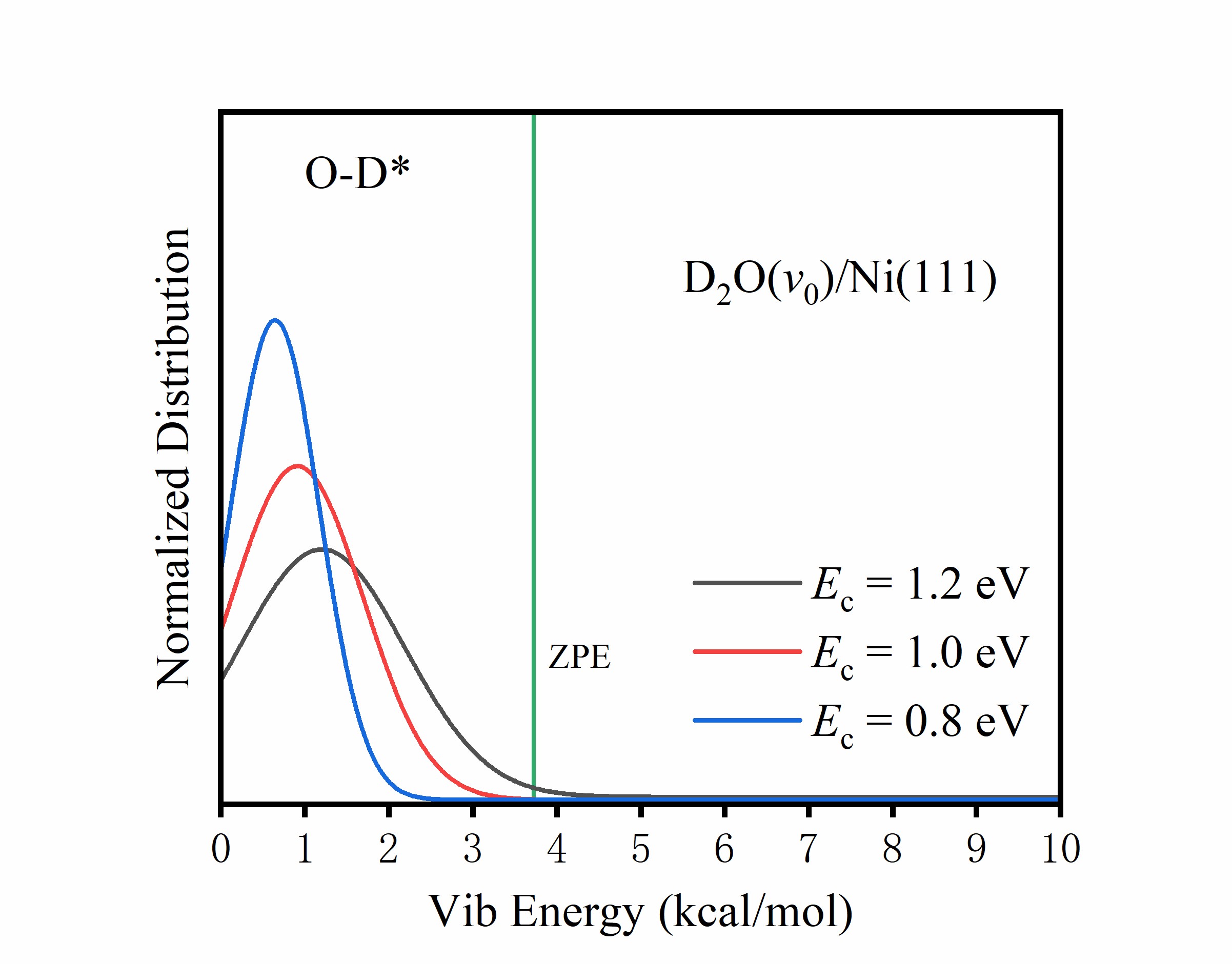
A practical quasi-classical trajectory method to avoid zero-point energy leakage in dissociative chemisorption of polyatomic molecules on surfaces
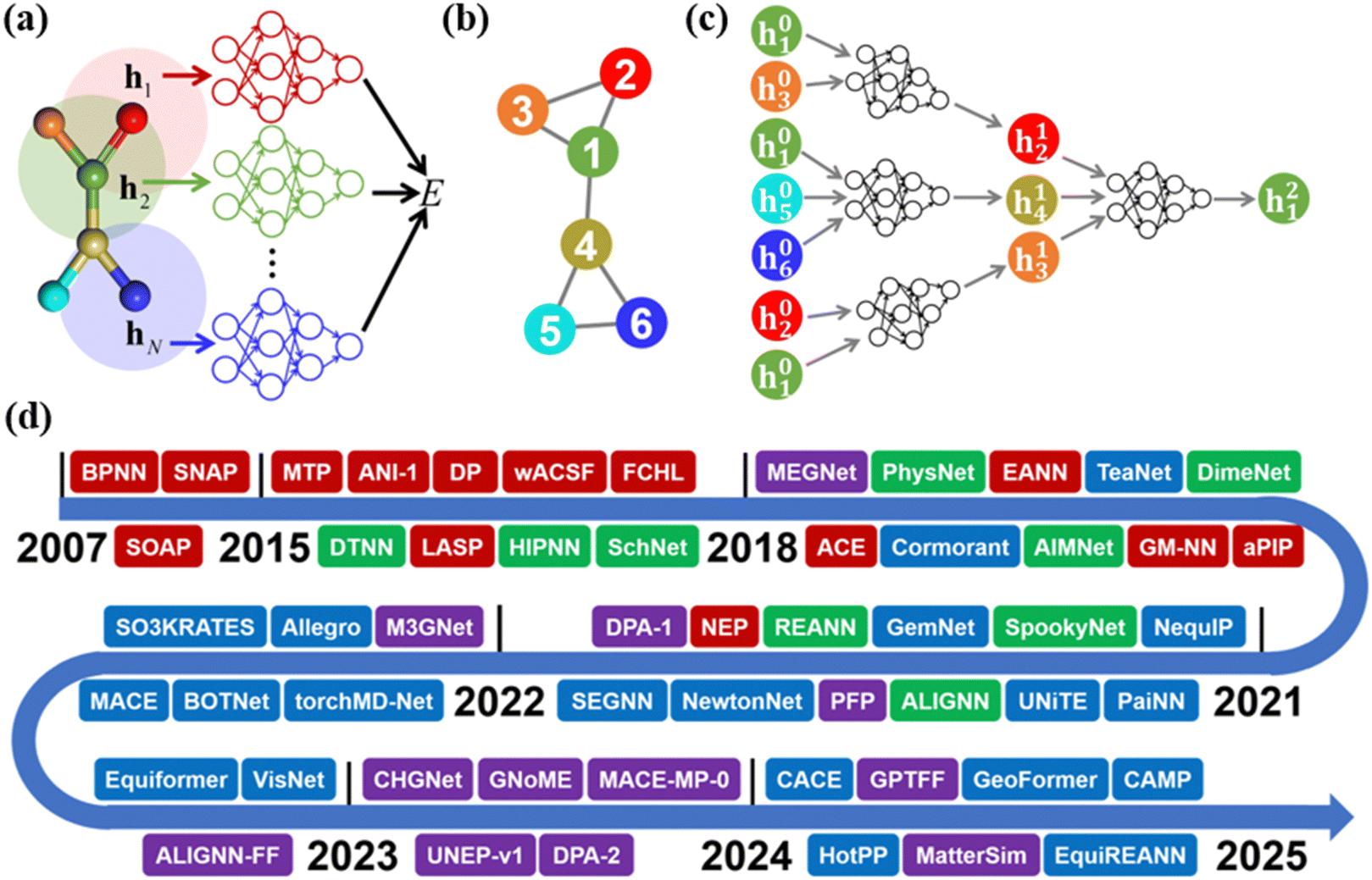
The evolution of machine learning potentials for molecules, reactions and materials
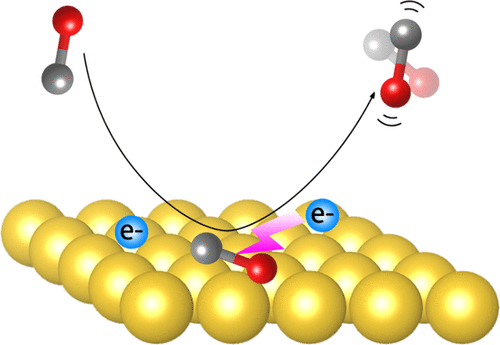
Vibrational Excitation in Gas-Surface Collisions of CO with Au (111): A First-Principles Nonadiabatic Dynamics Study
SchrödingerNet: A Universal Neural Network Solver for the Schrödinger Equation
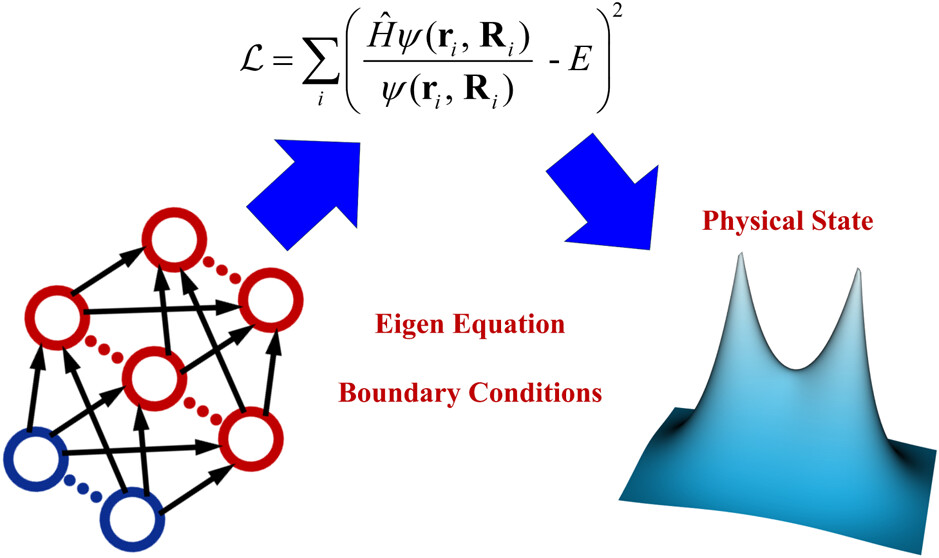
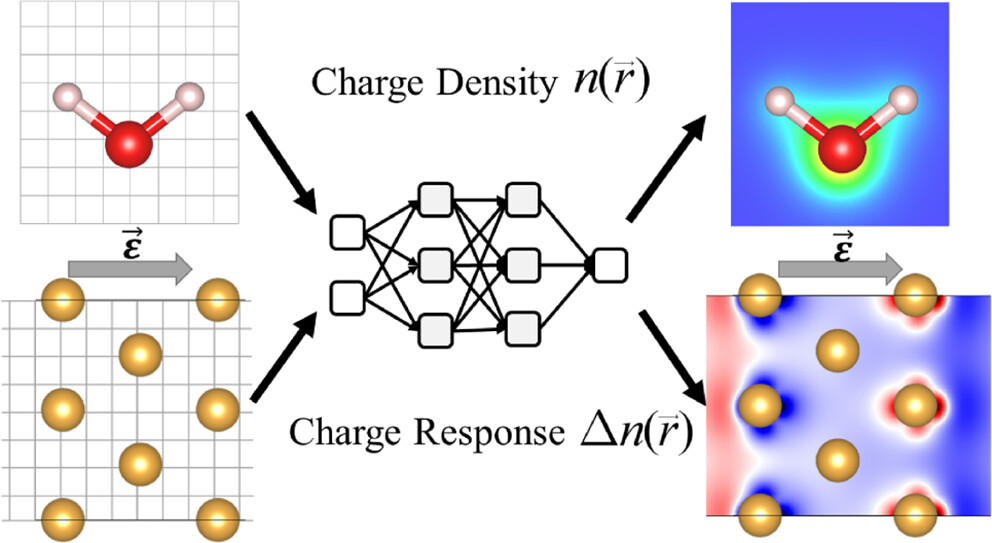
Efficient sampling for machine learning electron density and its response in real space
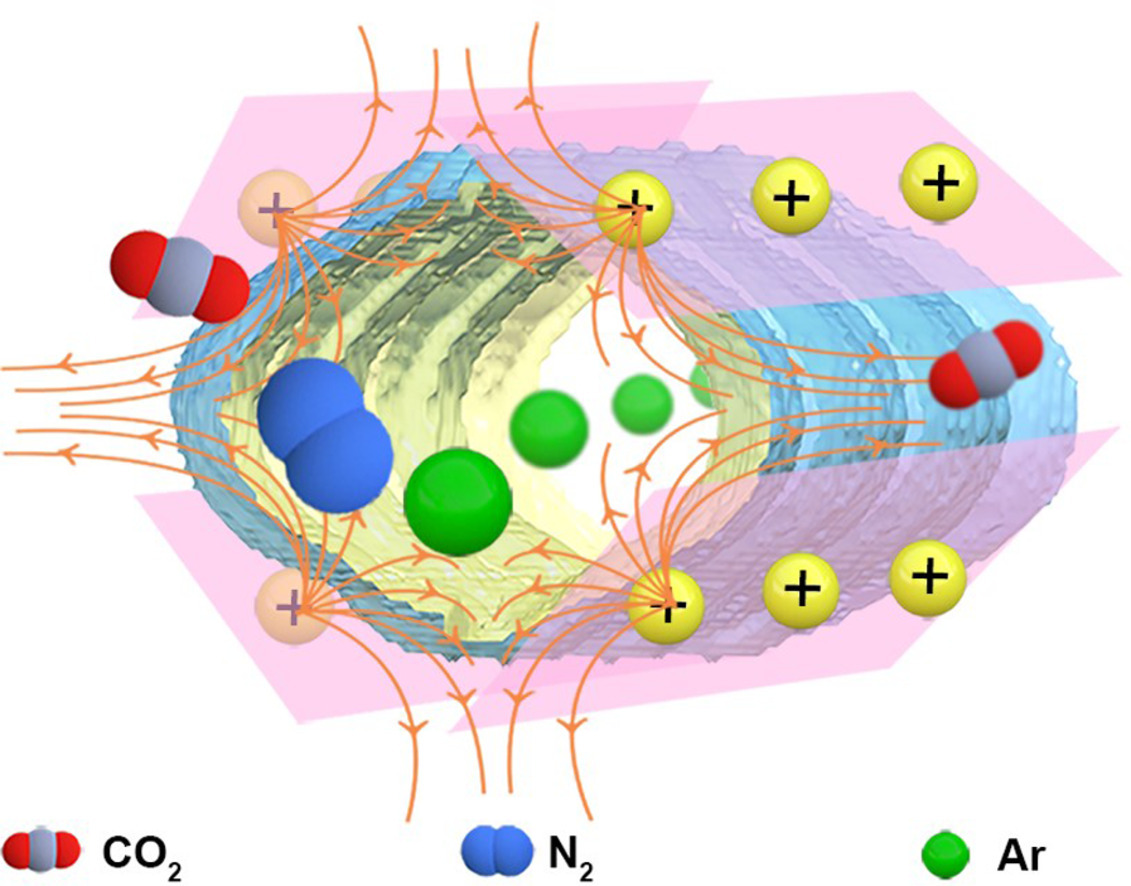
Confined electric field in nano-sized channels of ionic porous framework towards unique adsorption selectivity
Scattering at condensed-phase surfaces: general discussion
Mechanistic Insights into Nonadiabatic Interband Transitions on a Semiconductor Surface Induced by Hydrogen Atom Collisions
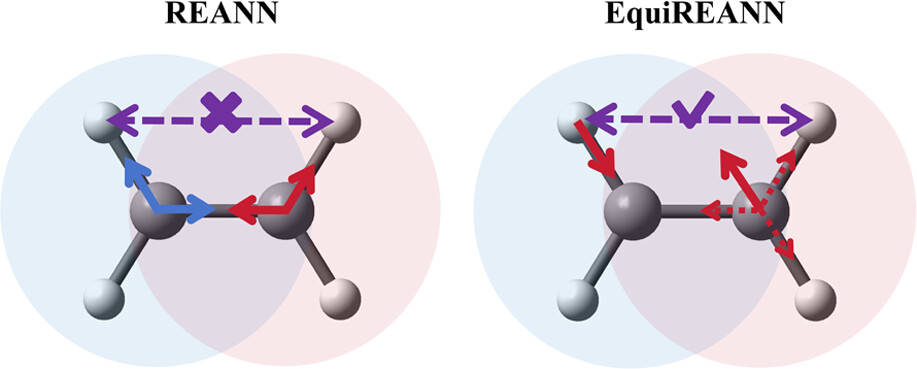
Simple and Efficient Equivariant Message-Passing Neural Network Model for Non-local Potential Energy Surfaces
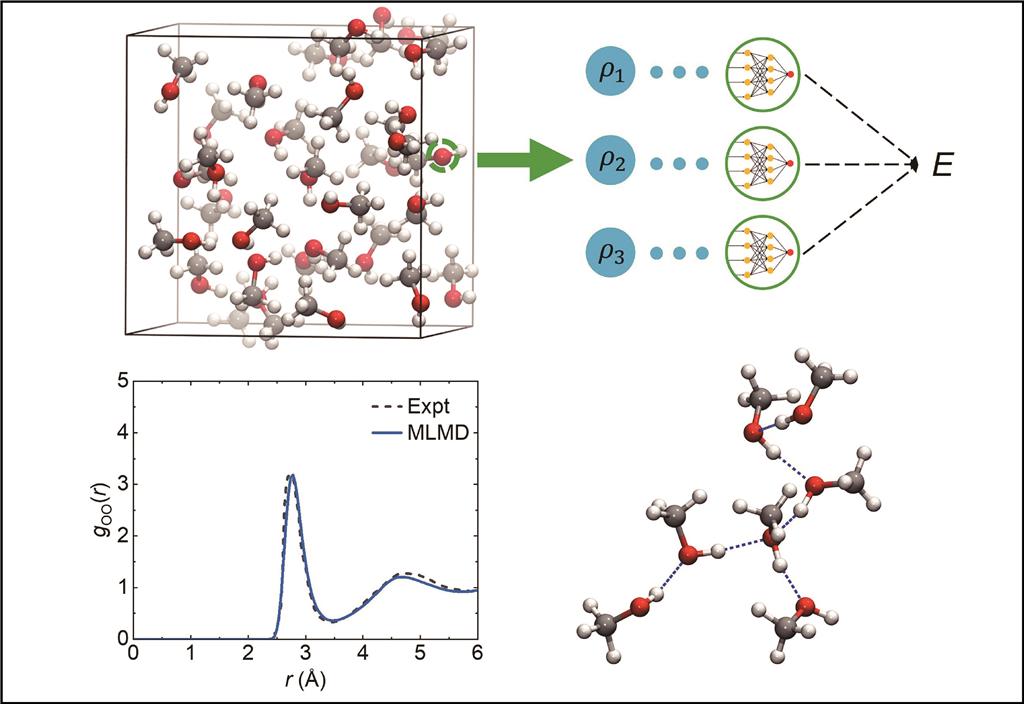
Machine learning molecular dynamics simulations of liquid methanol
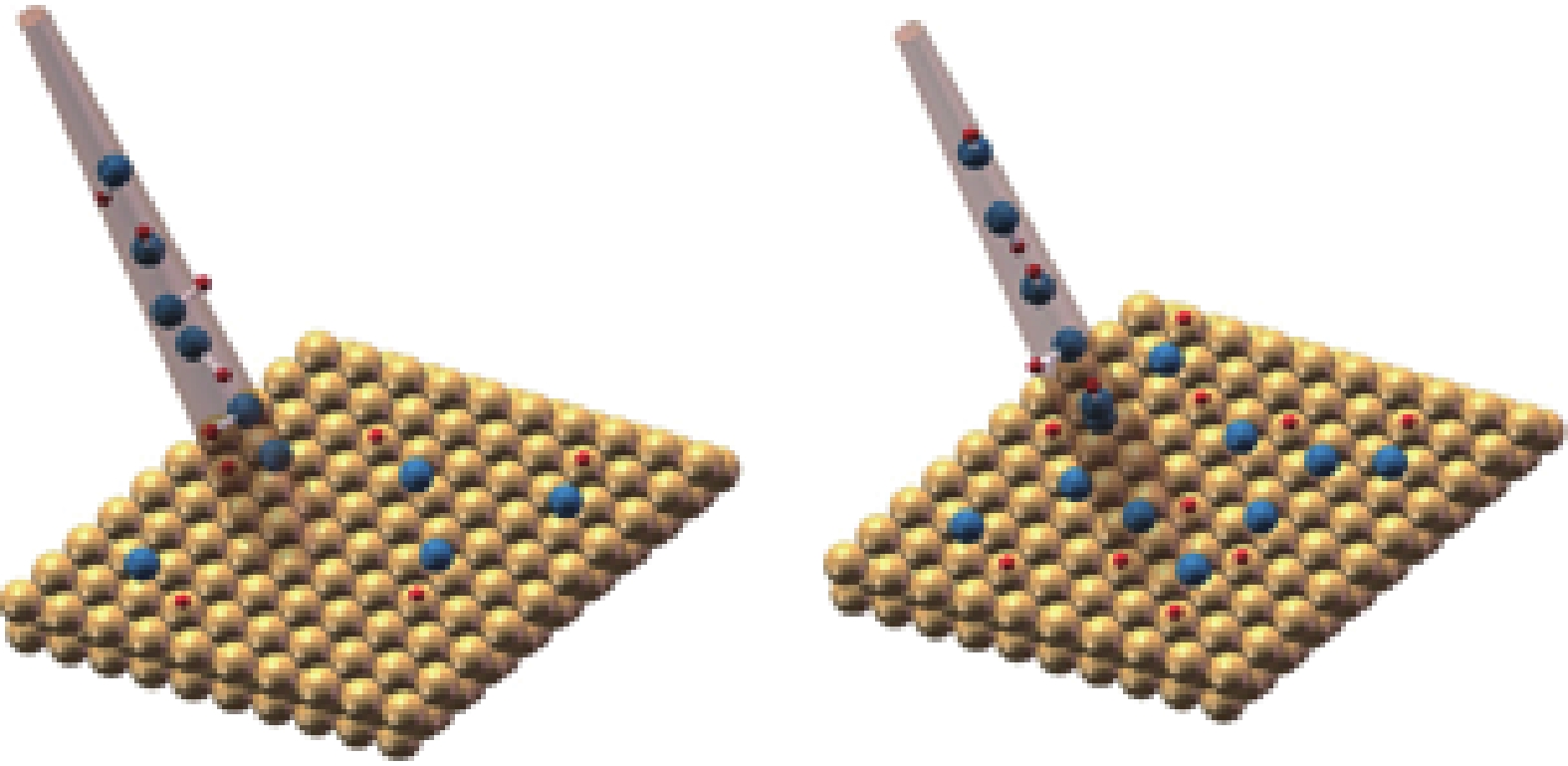
Coverage dependent dissociative adsorption of HCl on Au (111)
First-Principles Nonadiabatic Dynamics of Molecules at Metal Surfaces with Vibrationally Coupled Electron Transfer
Toward Efficient and Unified Treatment of Static and Dynamic Correlations in Generalized Kohn–Sham Density Functional Theory

Size dependent lithium-ion conductivity of solid electrolytes in machine learning molecular dynamics simulations
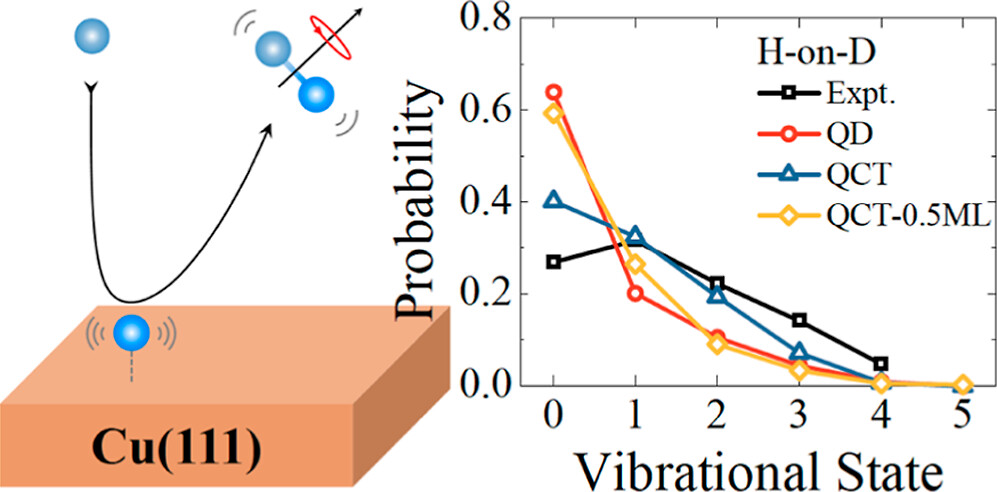
Comparison of Six-Dimensional Quantum and Quasi-Classical Dynamics of the Eley–Rideal Reaction of H (D) Atoms with D (H)-Covered Cu (111)
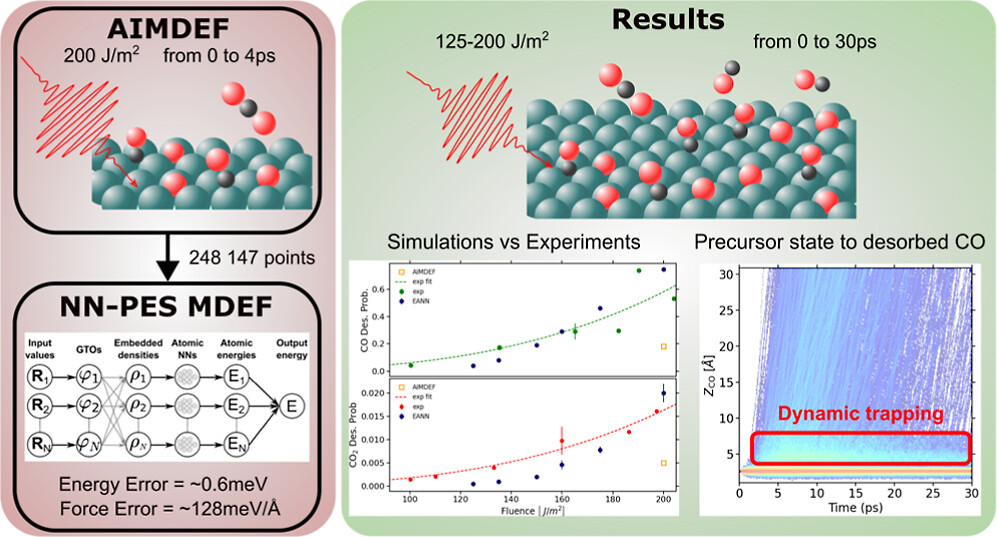
Understanding the Photoinduced Desorption and Oxidation of CO on Ru(0001) Using a Neural Network Potential Energy Surface
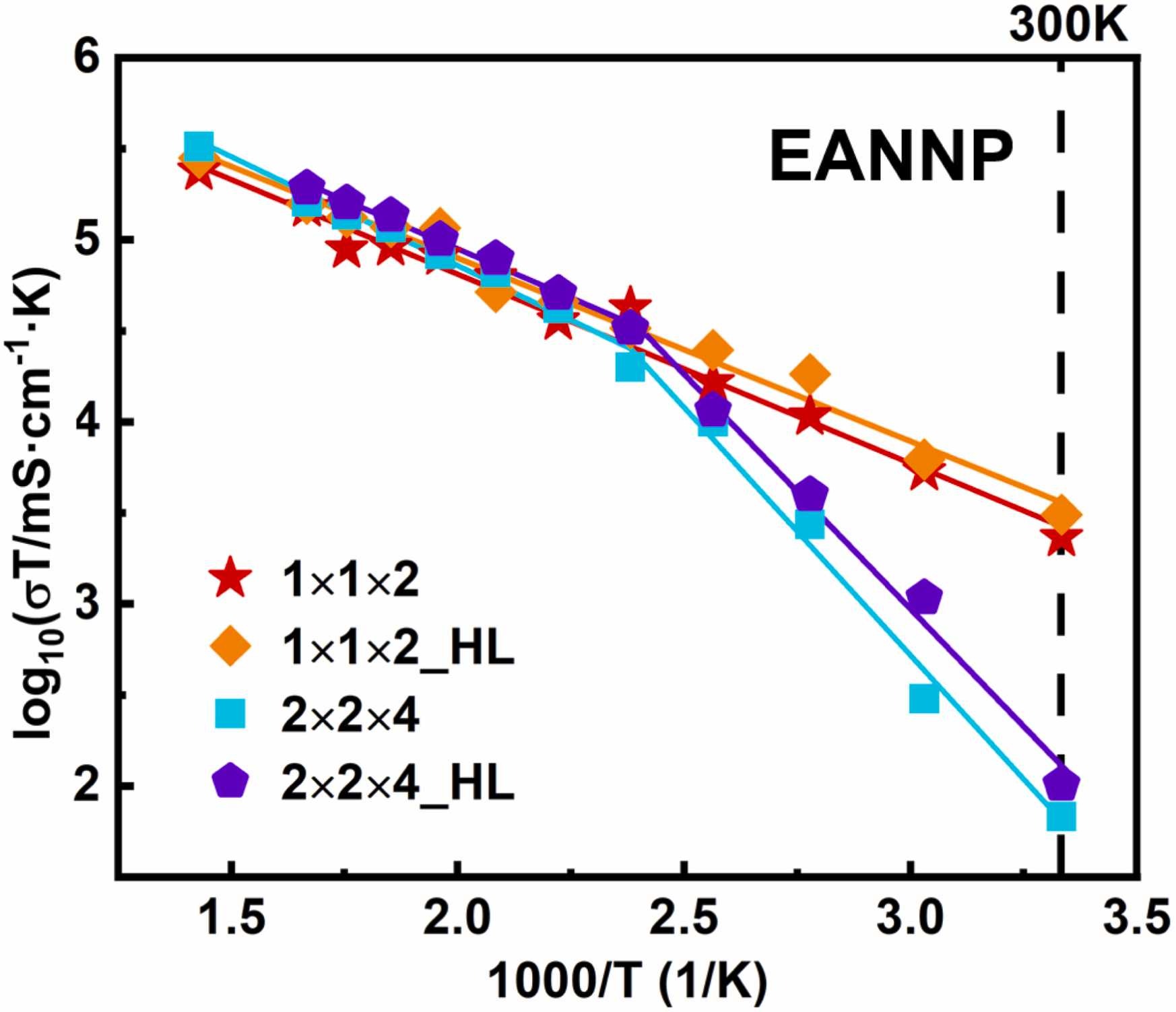
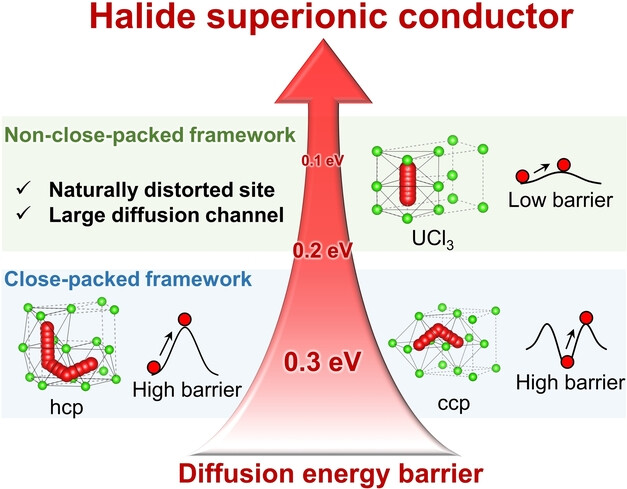
Halide Superionic Conductors with Non-Close-Packed Anion Frameworks
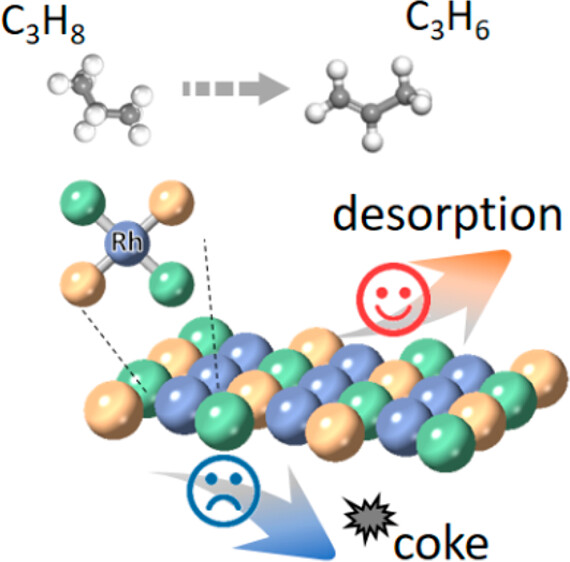
Phase-Transition-Induced Surface Reconstruction of Rh1 Site in Intermetallic Alloy for Propane Dehydrogenation
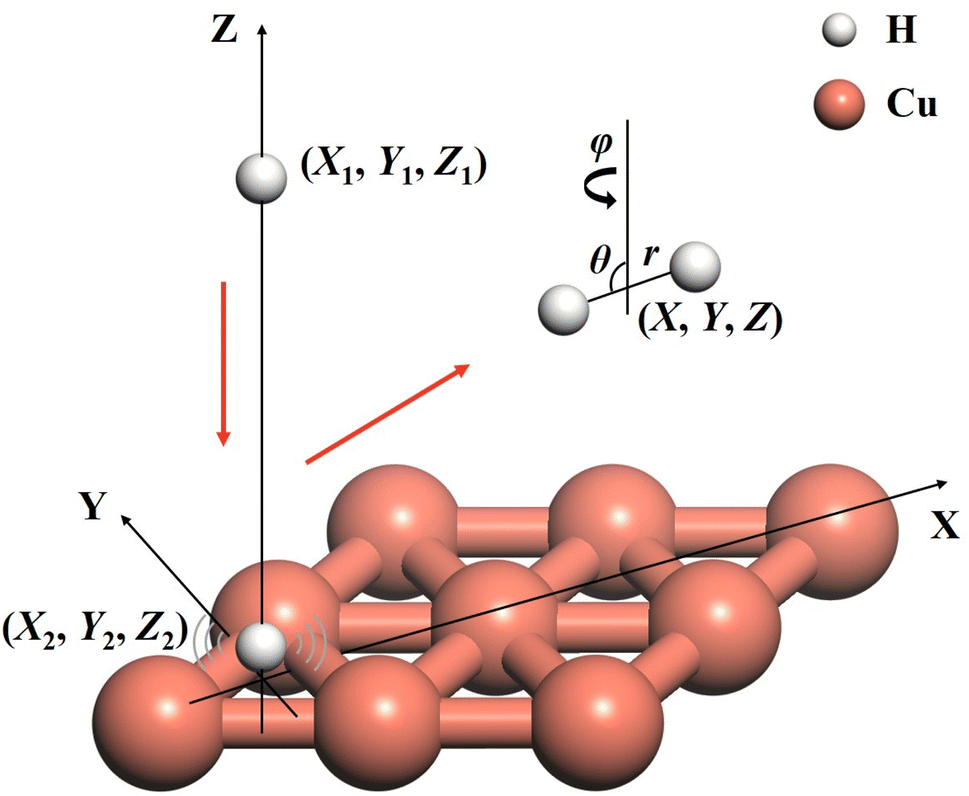
Six-dimensional quantum dynamics of an Eley–Rideal reaction between gaseous and adsorbed hydrogen atoms on Cu(111)
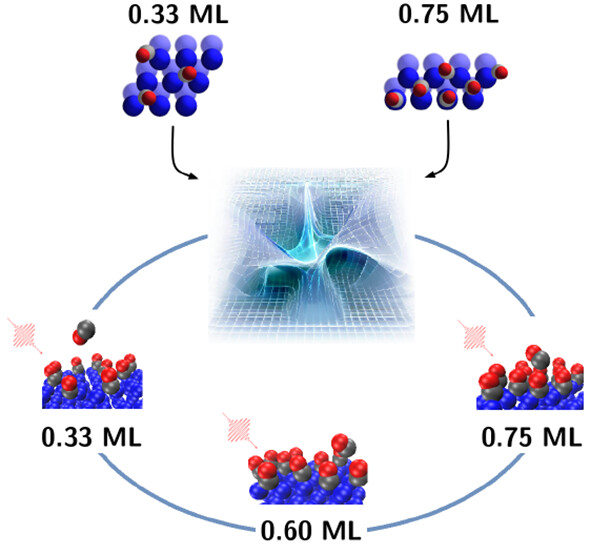
Multicoverage Study of Femtosecond Laser-Induced Desorption of CO from Pd(111)
Isotope effect suggests site-specific nonadiabaticity on Ge(111)c(2×8)
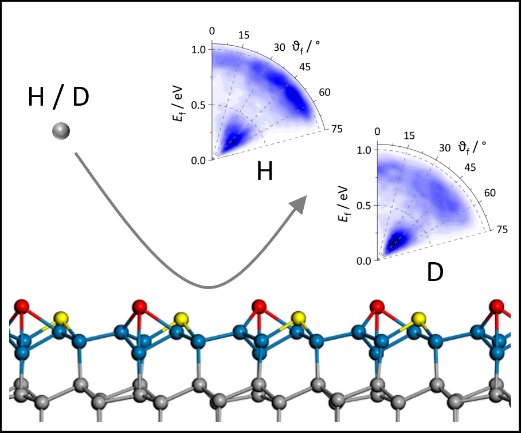
Dynamics of Collision-induced Energy Transfer
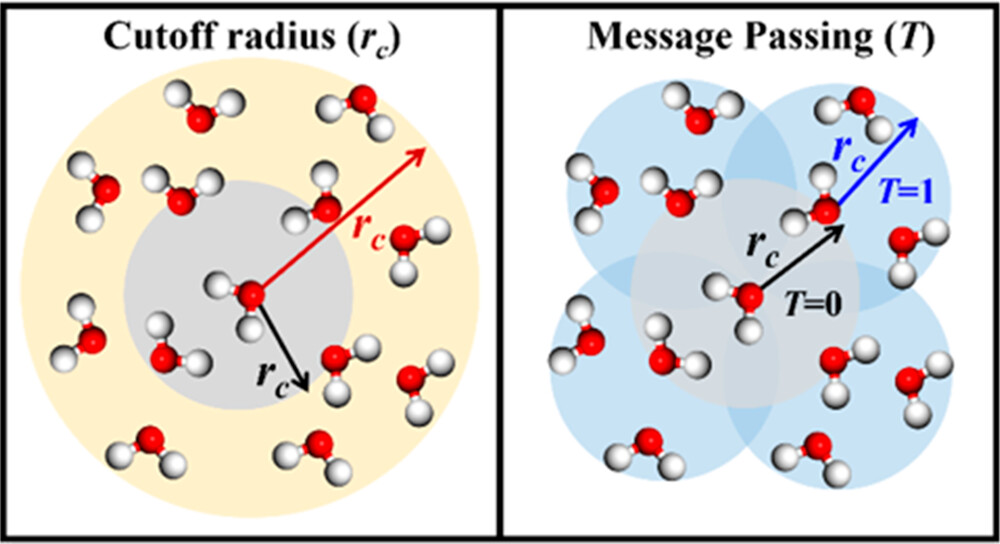
Accuracy Assessment of Atomistic Neural Network Potentials: The Impact of Cutoff Radius and Message Passing
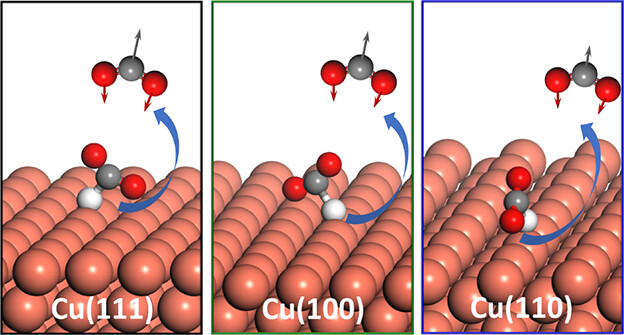
Theoretical Insights into Structure Sensitivity in Formate Decomposition Dynamics on Cu Surfaces
Universal machine learning for the response of atomistic systems to external fields
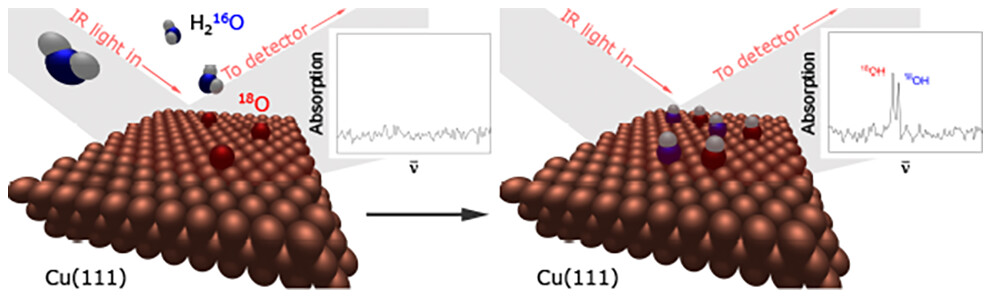
Probing Water Dissociation and Oxygen Replacement on Partially Oxygen-Covered Cu(111) by Reflection Absorption Infrared Spectroscopy
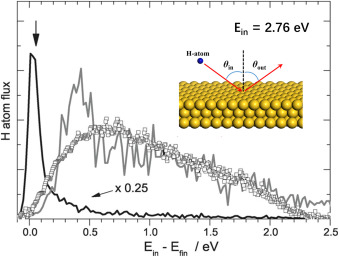
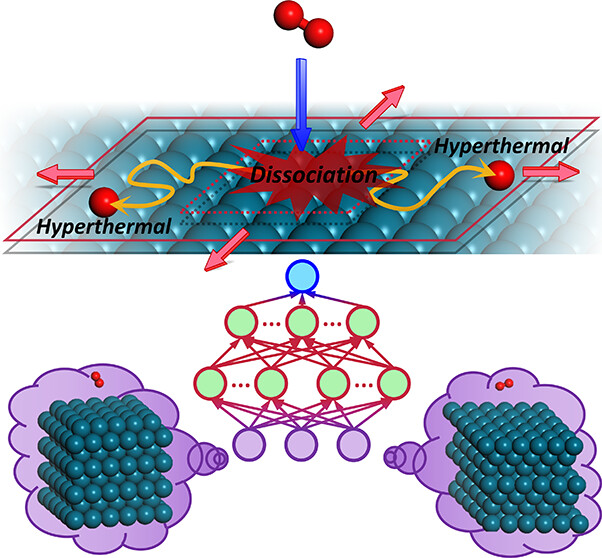
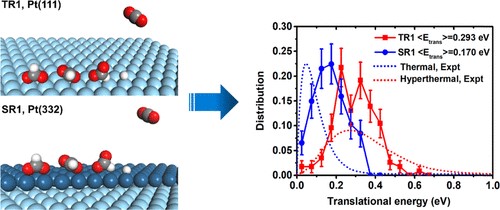
Modeling Equilibration Dynamics of Hyperthermal Products of Surface Reactions Using Scalable Neural Network Potential with First-Principles Accuracy
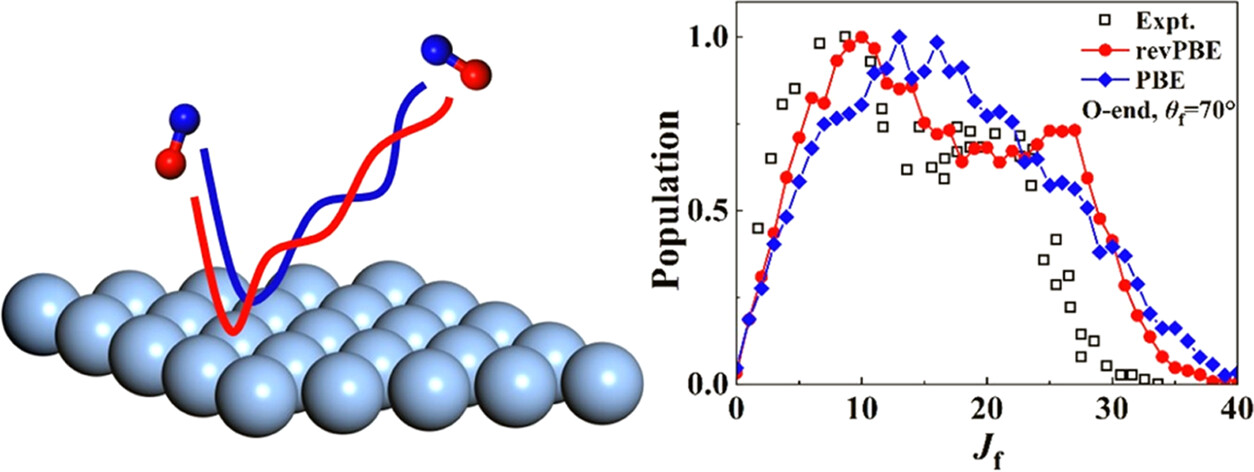
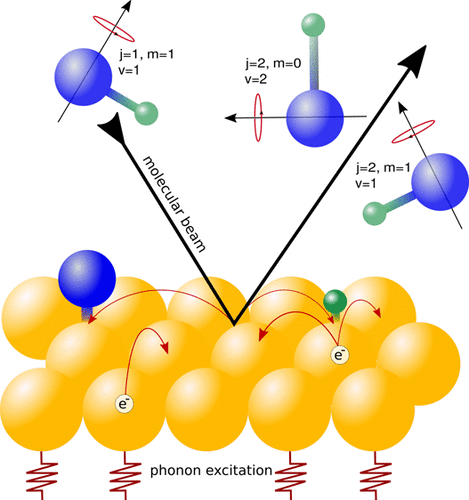
Rotationally Inelastic Scattering Dynamics of NO from Ag(111): Influence of Interaction Potentials
First-principles surface reaction rates by ring polymer molecular dynamics and neural network potential: role of anharmonicity and lattice motion
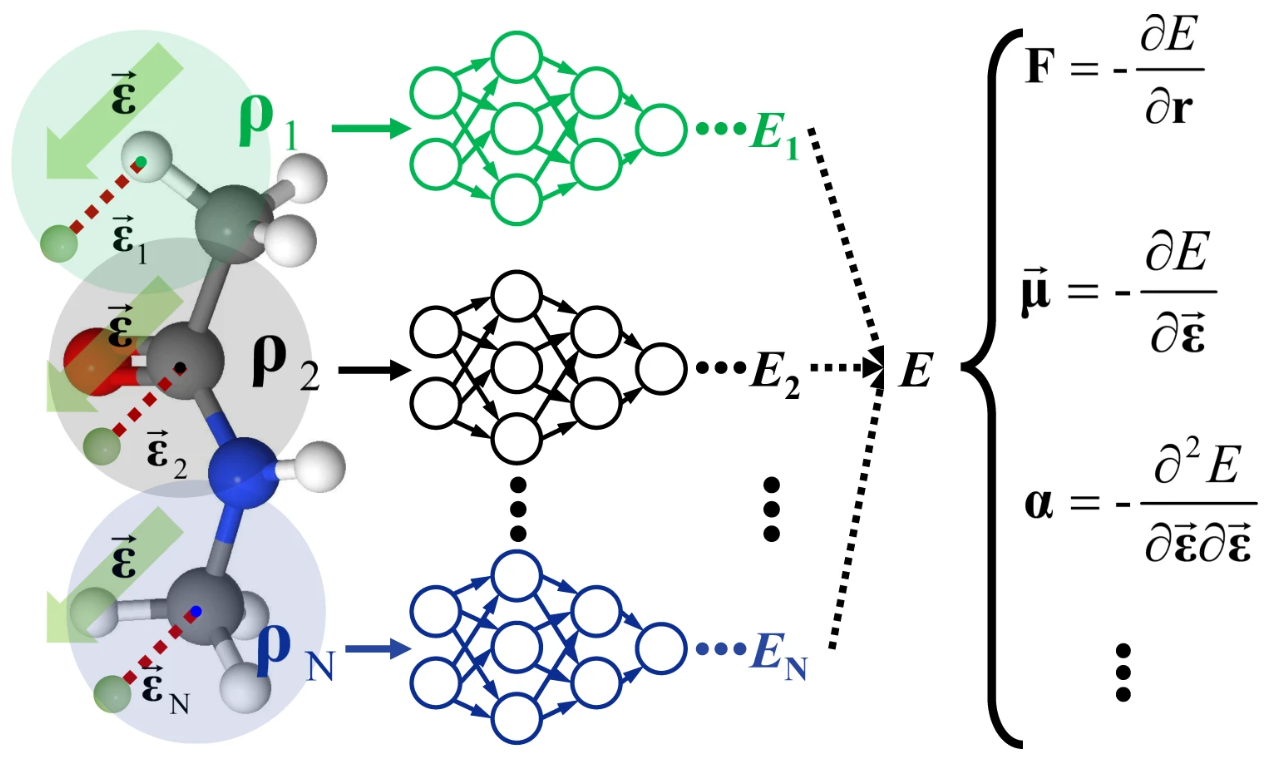
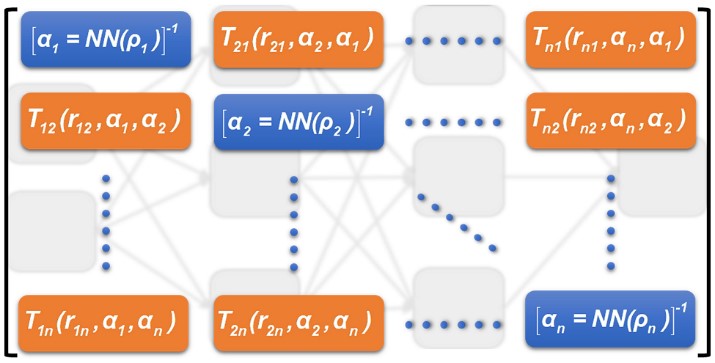
Chapter 19 - Learning dipole moments and polarizabilities
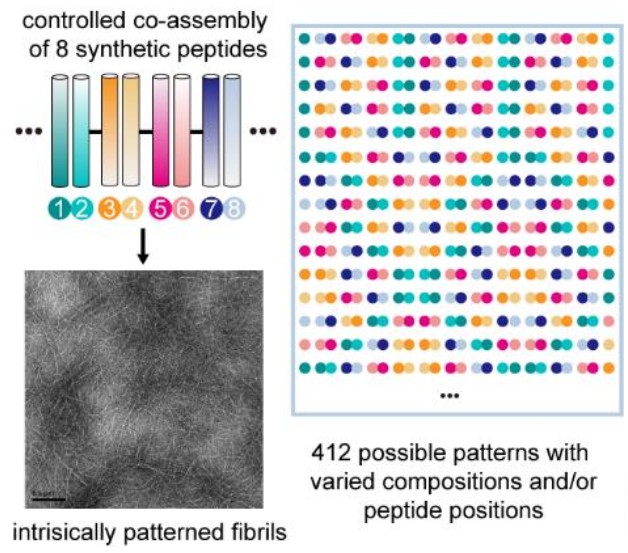
Design of Multicomponent Peptide Fibrils with Ordered and Programmable Compositional Patterns
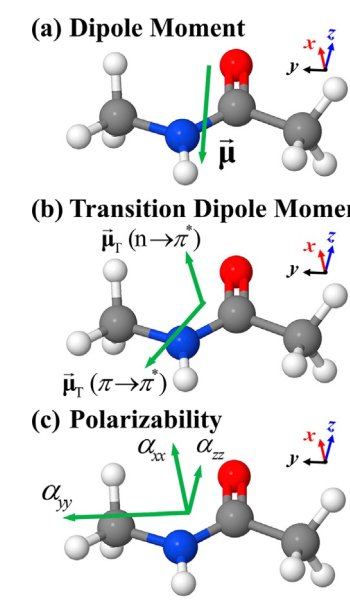
Accurate and Interpretable Dipole Interaction Model-Based Machine Learning for Molecular Polarizability
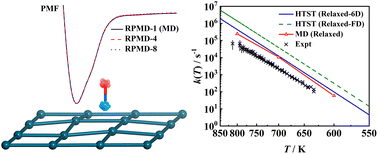
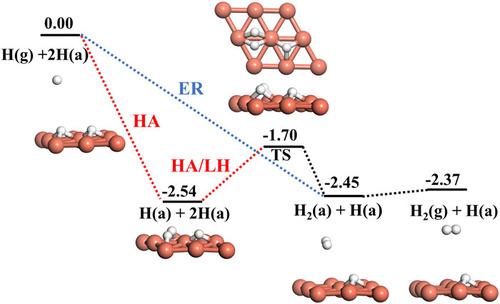
Investigating the Eley–Rideal recombination of hydrogen atoms on Cu (111) via a high-dimensional neural network potential energy surface
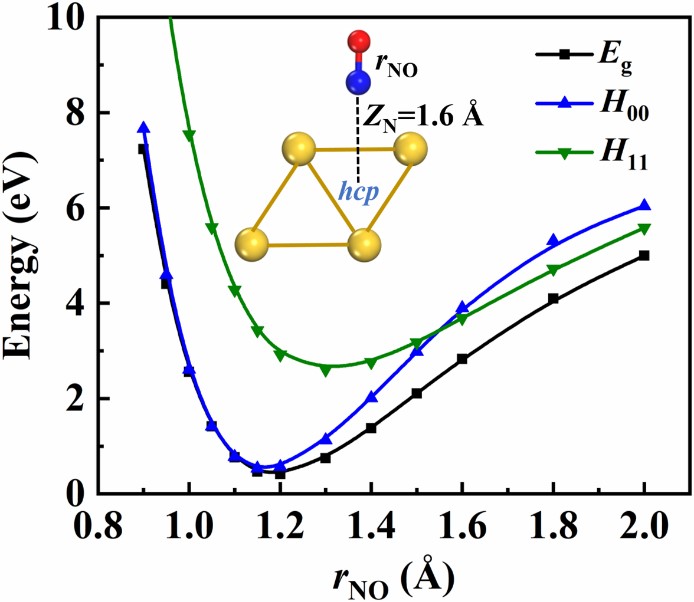
A pragmatic protocol for determining charge transfer states of molecules at metal surfaces by constrained density functional theory
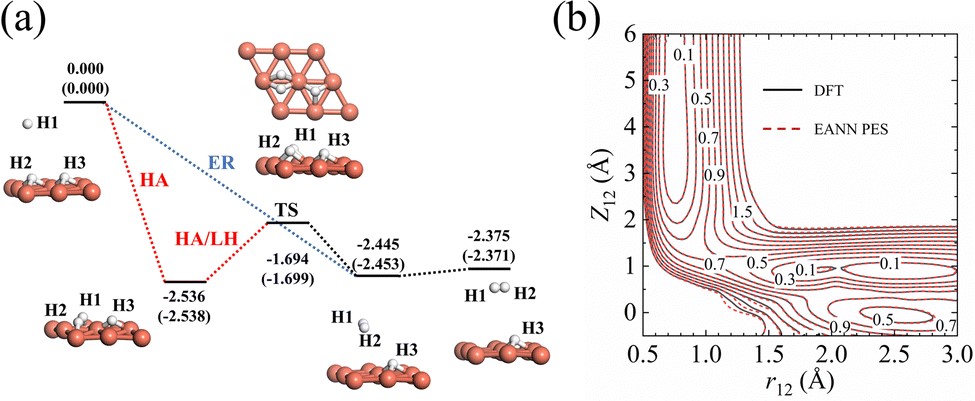
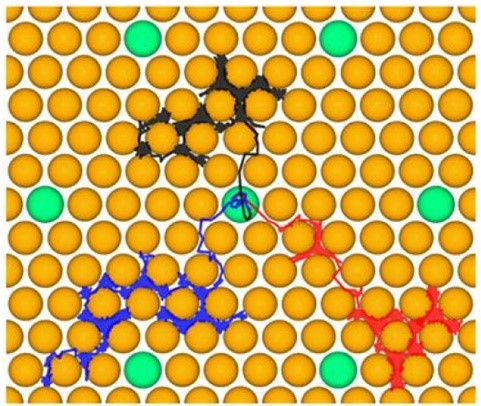
Short- and Long-Time Dynamics of Hydrogen Spillover from a Single Atom Platinum Active Site to the Cu(111) Host Surface
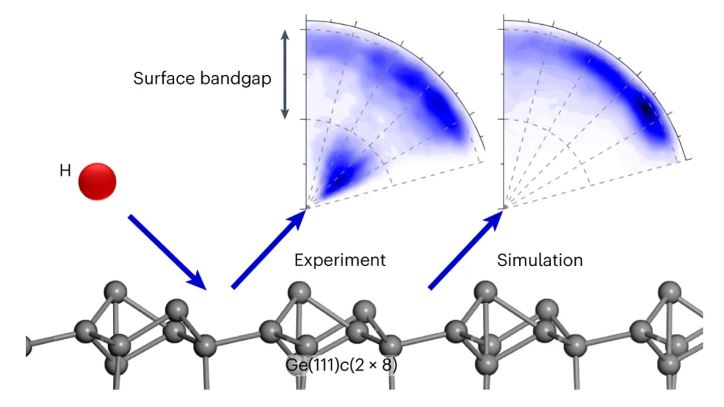
Hydrogen atom collisions with a semiconductor efficiently promote electrons to the conduction band
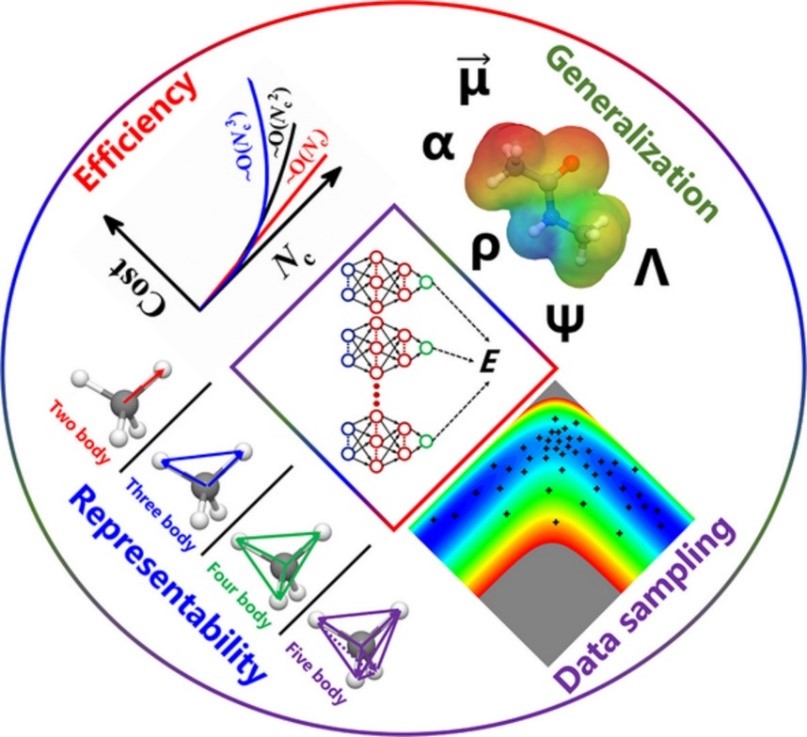
Atomistic neural network representations for chemical dynamics simulations of molecular, condensed phase, and interfacial systems: Efficiency, representability, and generalization
Stereodynamics of Adiabatic and Non-adiabatic Energy Transfer in a Molecule Surface Encounter
Vibrational Relaxation of Highly Vibrationally Excited Molecules Scattered from Au(111): Role of the Dissociation Barrier
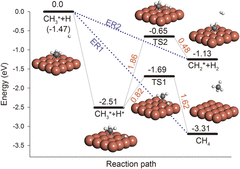
Mechanism and Dynamics of CO2 Formation in Formic Acid Decomposition on Pt Surfaces
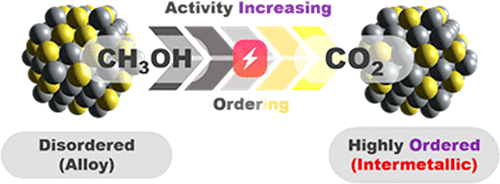
Ordering Degree-Dependent Activity of Pt3M (M = Fe, Mn) Intermetallic Nanoparticles for Electrocatalytic Methanol Oxidation
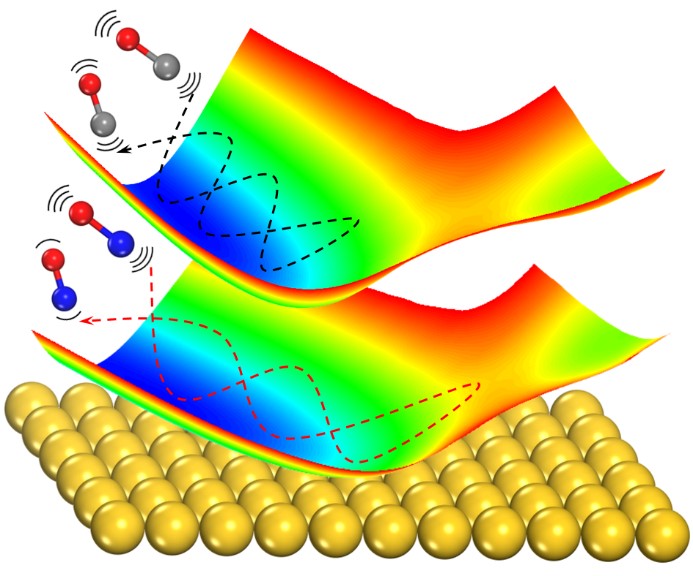
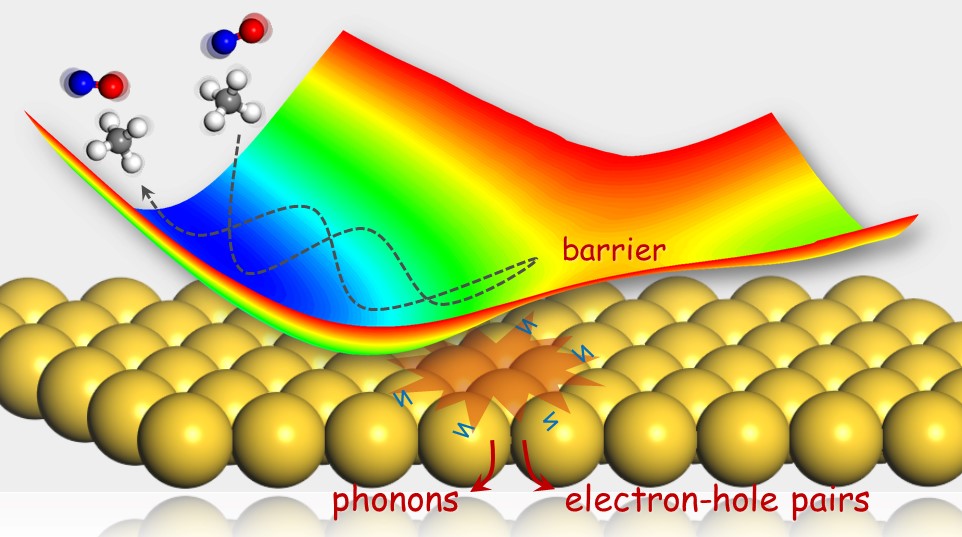
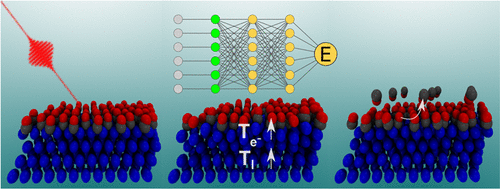
First-Principles Insights into Adiabatic and Nonadiabatic Vibrational Energy-Transfer Dynamics during Molecular Scattering from Metal Surfaces: The Importance of Surface Reactivity
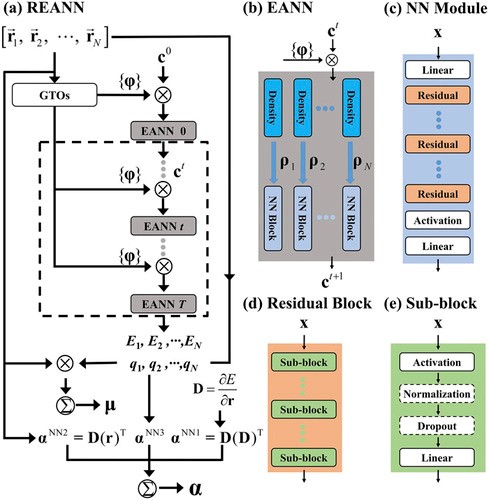
REANN: A PyTorch-based End-to-End Multi-functional Deep Neural Network Package for Molecular, Reactive and Periodic Systems
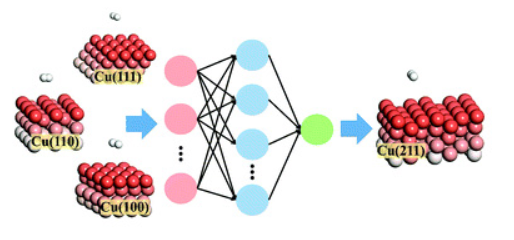
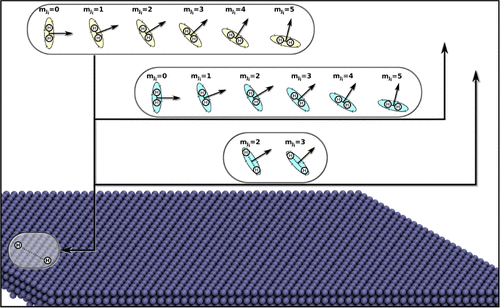
Six-dimensional State-to-State Quantum Dynamics of H2/D2 Scattering from Cu(100): Validity of the Site-Averaging Model
.gif)
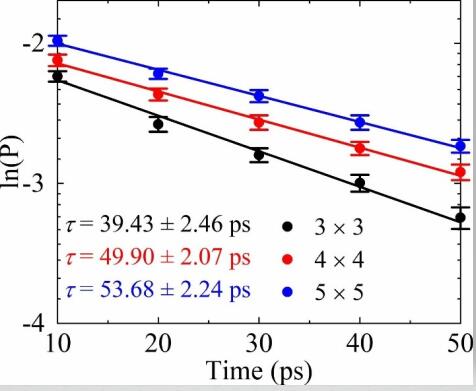
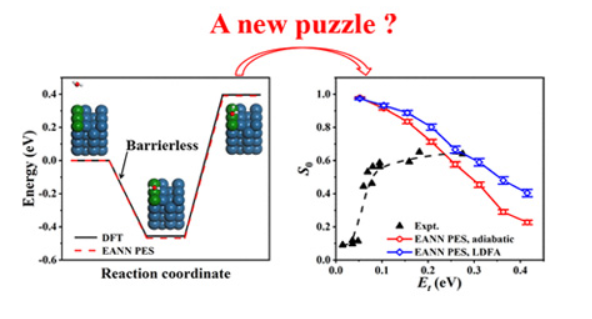
Influence of supercell size on Gas-Surface Scattering: A case study of CO scattering from Au(111)
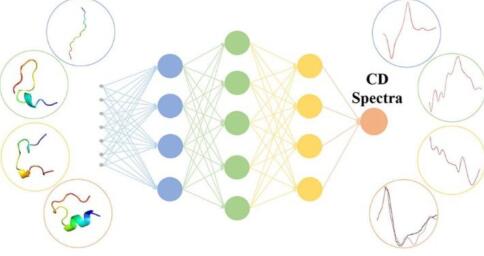
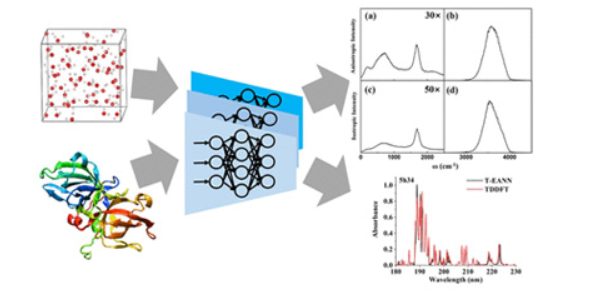
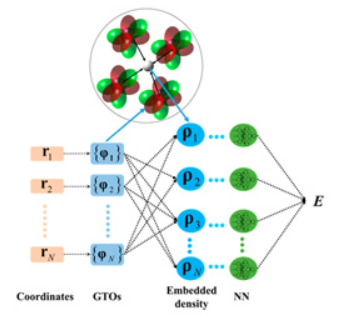
Accurate Machine Learning Prediction of Protein Circular Dichroism Spectra with Embedded Density Descriptors
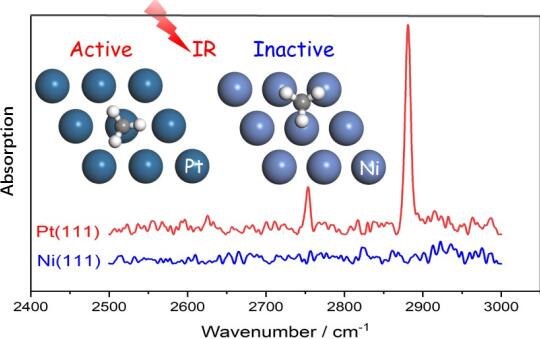
Infrared Activities of Adsorbed Species on Metal Surfaces: The Puzzle of Adsorbed Methyl (CH3)
Theoretical Study of Weakly Bound Adsorbates on Au(111): Tests on van der Waals Density Functionals

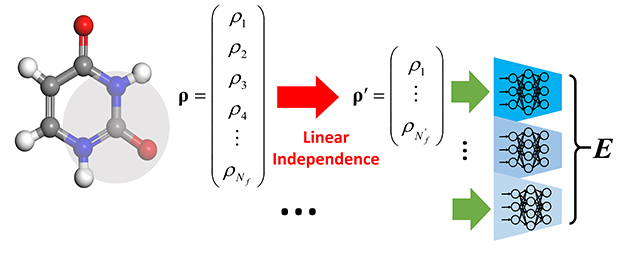
Efficient Selection of Linearly Independent Atomic Features for Accurate Machine Learning Potentials
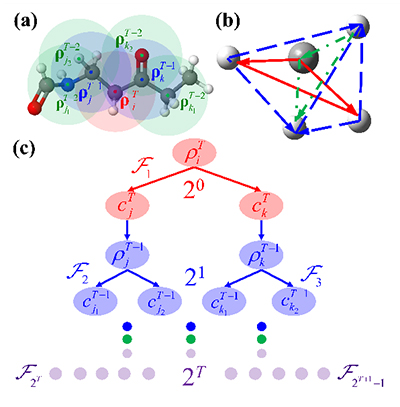
Physically Motivated Recursively Embedded Atom Neural Networks: Incorporating Local Completeness and Nonlocality
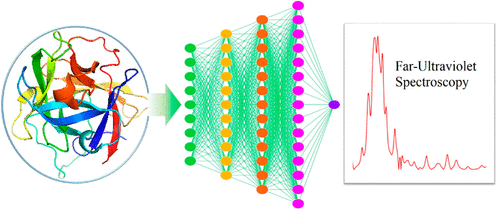
A Machine-Learning Protocol for Ultraviolet Protein-Backbone Absorption Spectroscopy under Environmental Fluctuations
Photoinduced Desorption Dynamics of CO from Pd(111): A Neural Network Approach
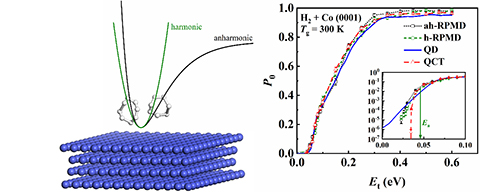
Ring Polymer Molecular Dynamics in Gas-Surface Reactions: Tests on Initial Sampling and Potential Energy Landscape
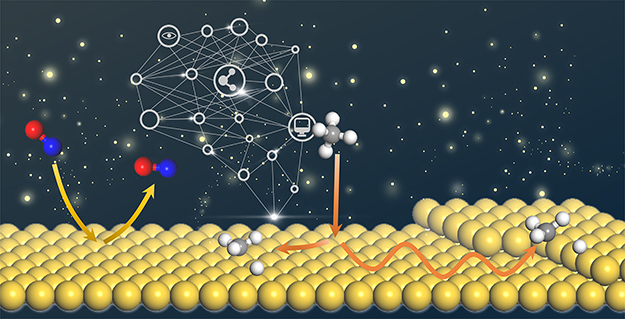
Neural Network Representations for Studying Gas-Surface Reaction Dynamics: Beyond the Born-Oppenheimer Static Surface Approximation
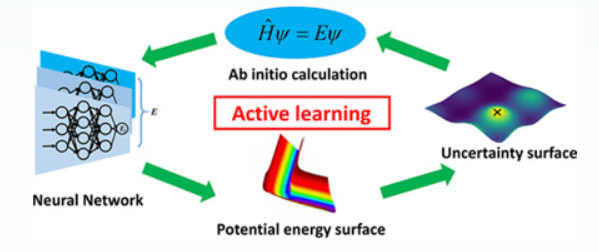
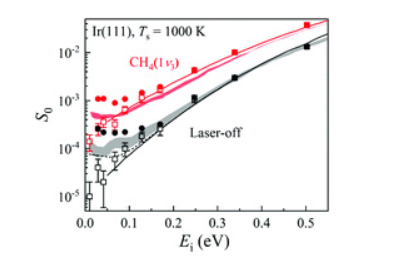
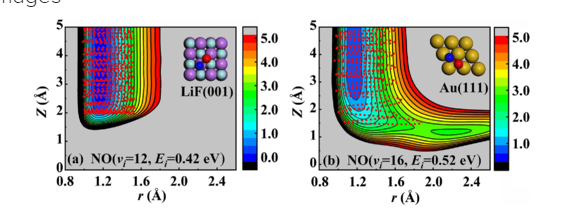
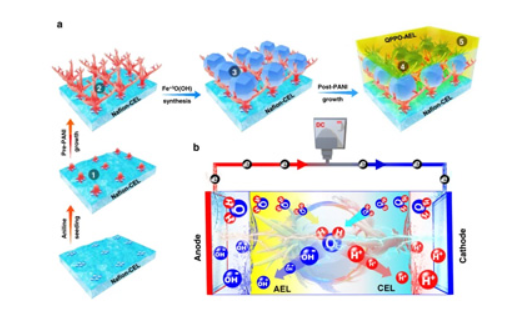
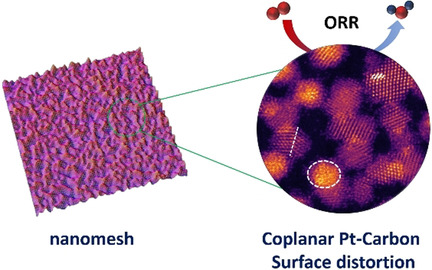
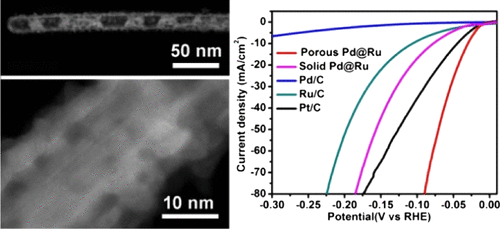
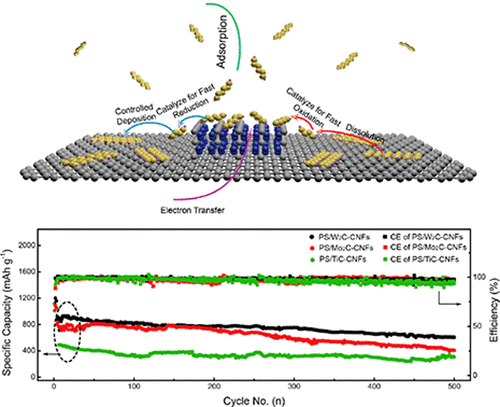
Coplanar Pt/C Nanomeshes with Ultrastable Oxygen Reduction Performance in Fuel Cells
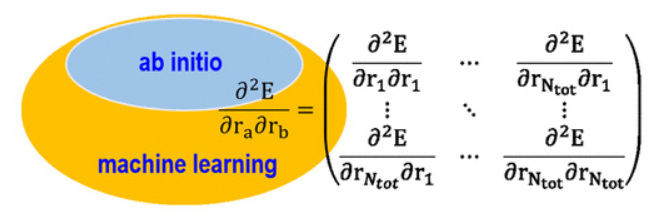
Efficient Construction of Excited-State Hessian Matrices with Machine Learning Accelerated MultiLayer Energy-Based Fragment Method
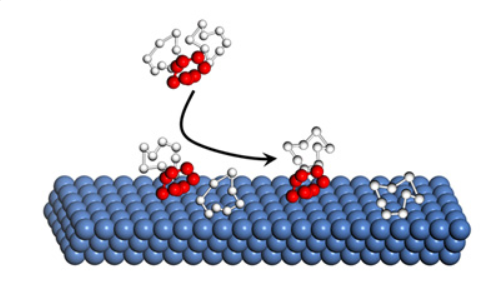
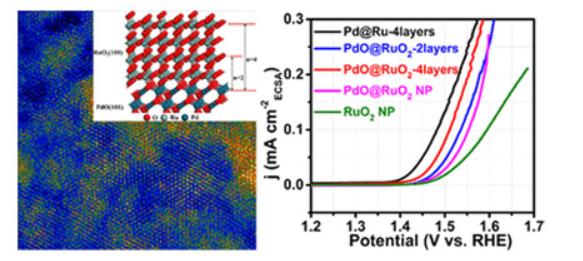
Engineering the Atomic Layer of RuO2 on PdO Nanosheets Boosts Oxygen Evolution Catalysis
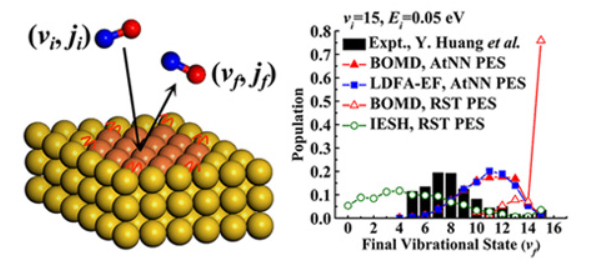
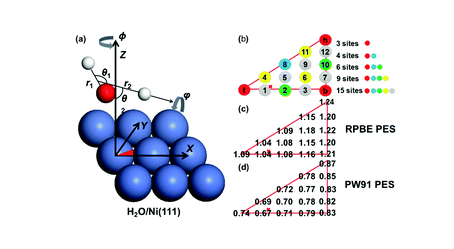
Efficient Vibrational Energy Redistribution between Stretching Modes: State-to-State Quantum Scattering of H2O from Cu(111)
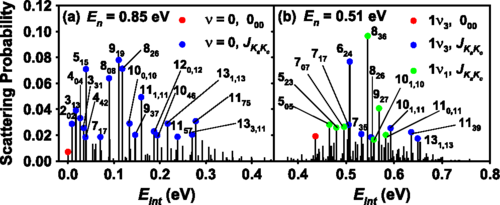
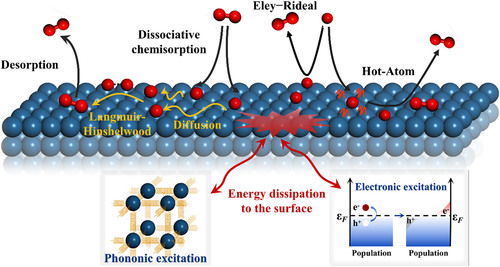
Reactive and Nonreactive Scattering of HCl from Au(111): An Ab Initio Molecular Dynamics Study
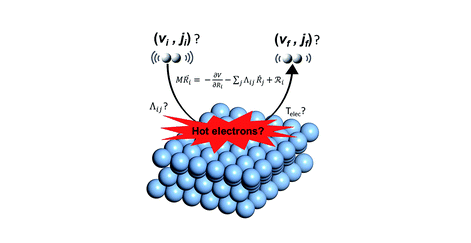
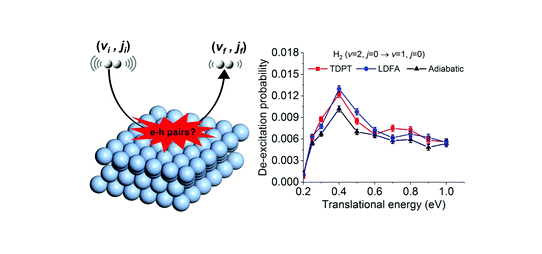
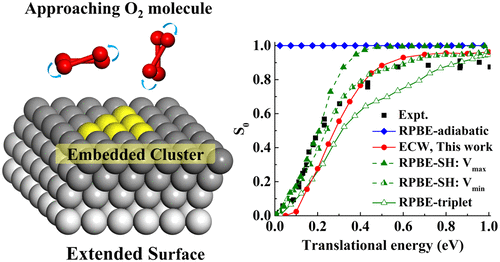
Dissociative Chemisorption of O2 on Al(111): Dynamics on a Correlated Wave-Function-Based Potential Energy Surface
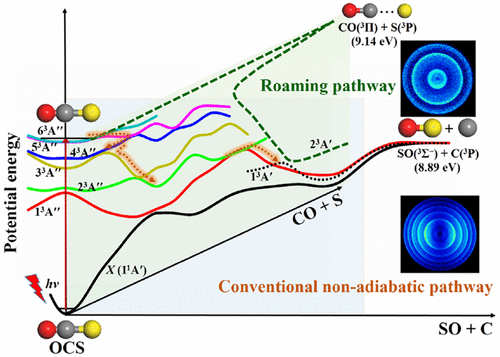
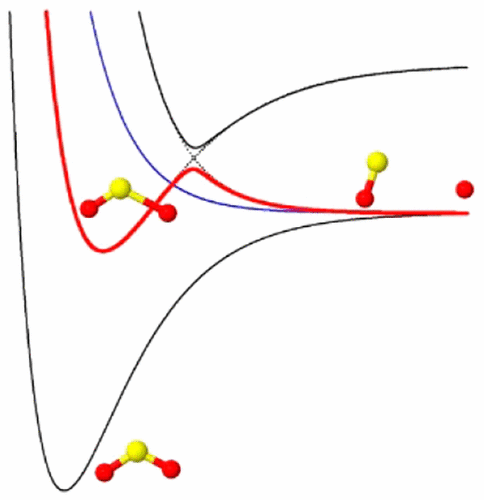
Final State Resolved Quantum Predissociation Dynamics of SO2(C̃1B2)and Its Isotopomers via a Crossing with a Singlet Repulsive State
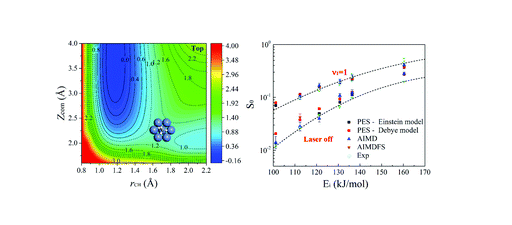
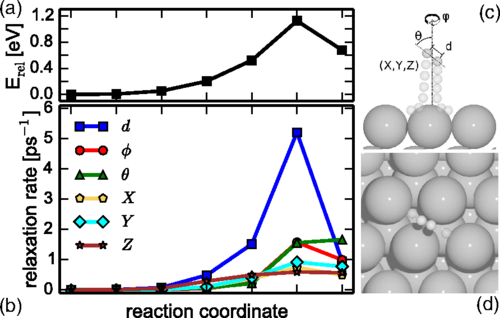
Mode Specific Electronic Friction in Dissociative Chemisorption on Metal Surfaces:H2 on Ag(111)
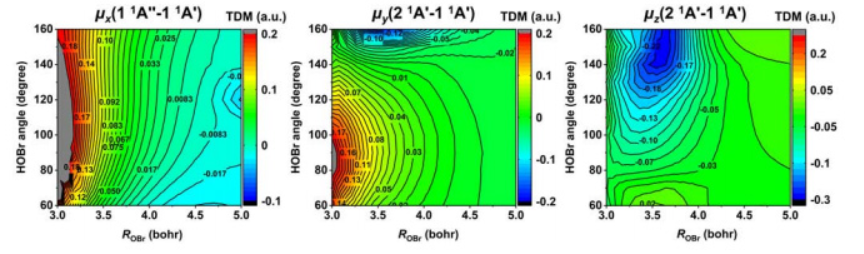
Photoabsorption Assignments for the C̃1B2← X̃1A1 Vibronic Transitions of SO2,Using New Ab Initio Potential Energy and Transition Dipole Surfaces
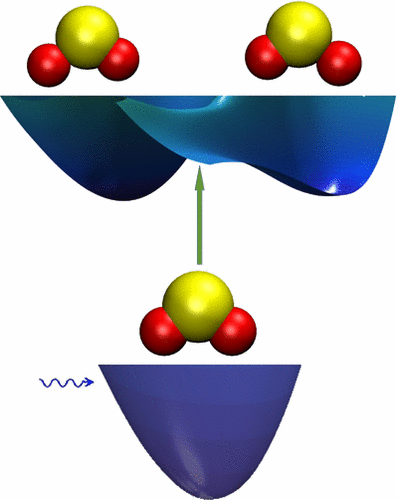
First-principles C band absorption spectra of SO2 and its isotopologues
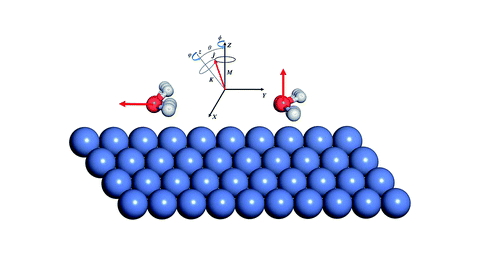
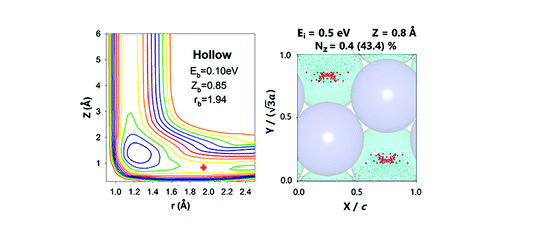
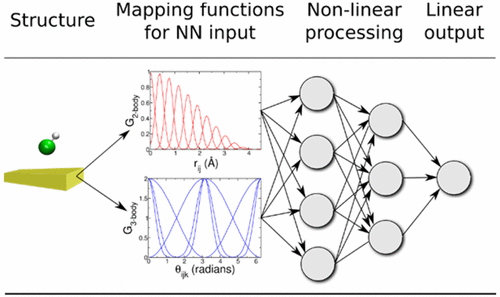
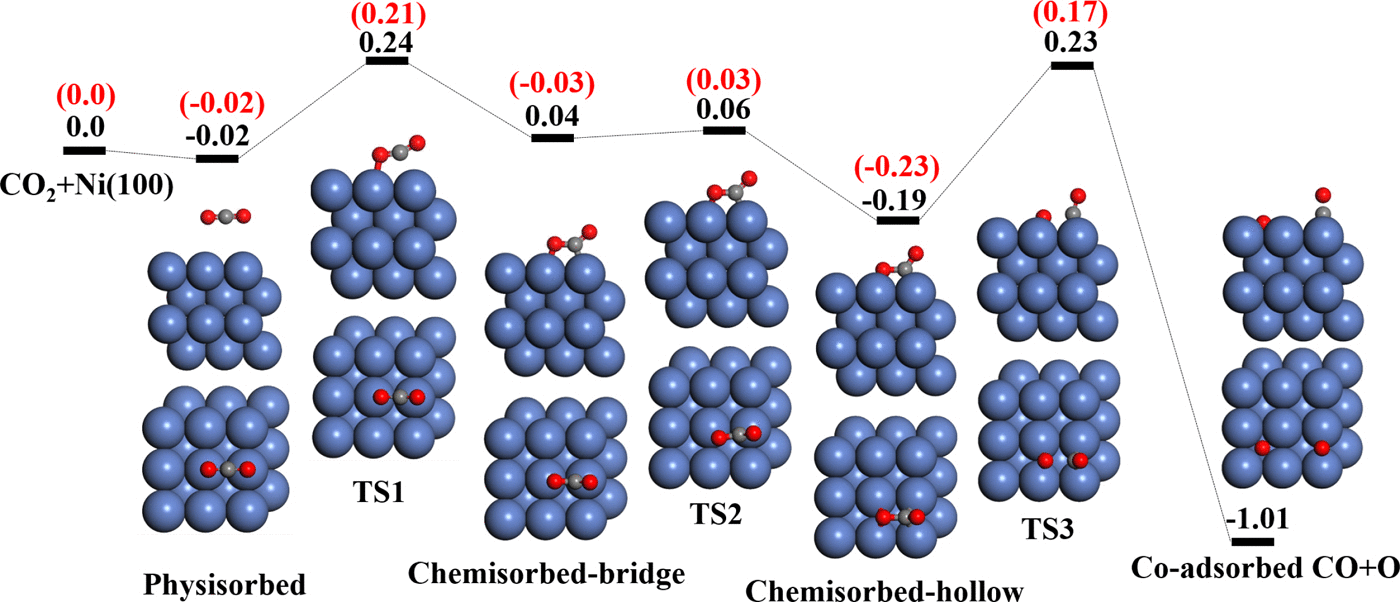
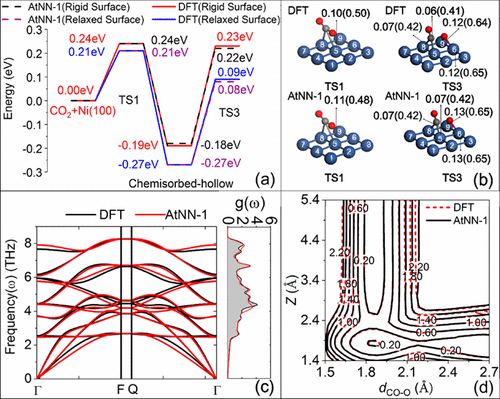
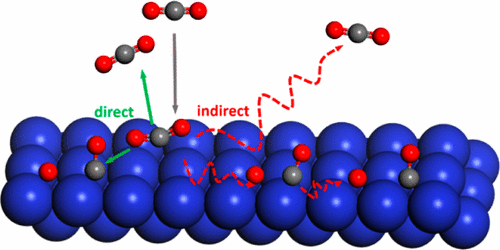
Ab Initio Molecular Dynamics Study of Dissociative Chemisorption and Scattering of CO2 on Ni(100): Reactivity, Energy Transfer, Steering Dynamics, and Lattice Effect
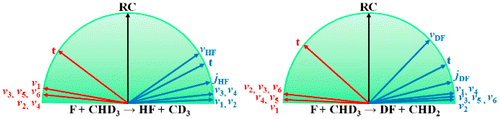
State-to-State Mode Specificity in F + CHD3 --> HF/DF + CD3/CHD2 Reaction
New ab initio adiabatic potential energy surfaces and bound state calculations for the singlet ground X̃1A1 and excited C̃1B2(21A′) states of SO2
Communication: Enhanced dissociative chemisorption of CO2 via vibrational excitation