Our research involves theory and computation for a better understanding of chemical reaction dynamics at the molecular level. We focus on elementary chemical reactions of small molecules on surfaces, which are related to many important interfacial processes, such as heterogeneous catalysis, material growth, and corrosion. It is indeed very challenging to reveal the most detailed information of these reactions for a quantitative comparison with the state-of-the-art experimental data with quantum state resolution. Our efforts include:
1. Machine learning representations for potential energy surface and other molecular properties
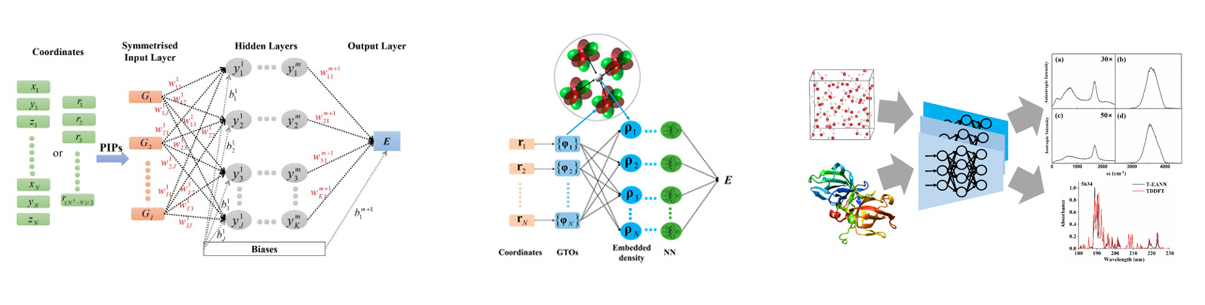
Potential energy surface (PES) is a natural consequence of Born-Oppenheimer approximation, which determines the motion of atoms and thus plays a central role in understanding chemical dynamics. We develop machine learning representations of highly-accurate multi-dimensional PESs. It is important to realize that the PES should be invariant with respect to the overall translation and rotation of the system, and permutation of identical atoms. The key is to make suitable descriptors of molecular (or material) configurations satisfying the symmetry requirement as machine learning programs themselves do not recognize chemical symmetry. For small systems, we proposed to use the permutationally invariant polynomial basis as the input of neural networks, yielding the so-called "PIP-NN" approach1, 2. For large systems, e.g. molecules interacting with the surface with many surface atoms, we proposed a physically inspired embedded atom neural network (EANN) approach3, 4, which uses the embedded electron density at each atomic position to represent the local environment surrounding this atom. By expressing the embedded density in terms of the square of linear combinations of neighboring atomic orbitals, we obtain a symmetry-preserving and efficient descriptor for atom-centered structure. We further extend the machine learning representation for permeant dipole, transition dipole, polarizability of molecules and materials5, as well as electronic friction tensor of molecules on surfaces6.
References:
1、B. Jiang, and H. Guo, J. Chem. Phys. 139, 054112 (2013).
2、B. Jiang, J. Li, and H. Guo, Int. Rev. Phys. Chem. 35, 479 (2016).
3、Y. Zhang, C. Hu, and B. Jiang, J. Phys. Chem. Lett. 10, 4962 (2019).
4、Y. Zhang, C. Hu, and B. Jiang, Phys. Chem. Chem. Phys. 23, 1815 (2021).
5、Y. Zhang, S. Ye, J. Zhang, C. Hu, J. Jiang, and B. Jiang, J. Phys. Chem. B 124, 7284 (2020).
6、Y. Zhang, R. J. Maurer, and B. Jiang, J. Phys. Chem. C 124, 186 (2020).
2. Mechanisms and dynamics of molecular energy conversion and chemical reactions at surfaces
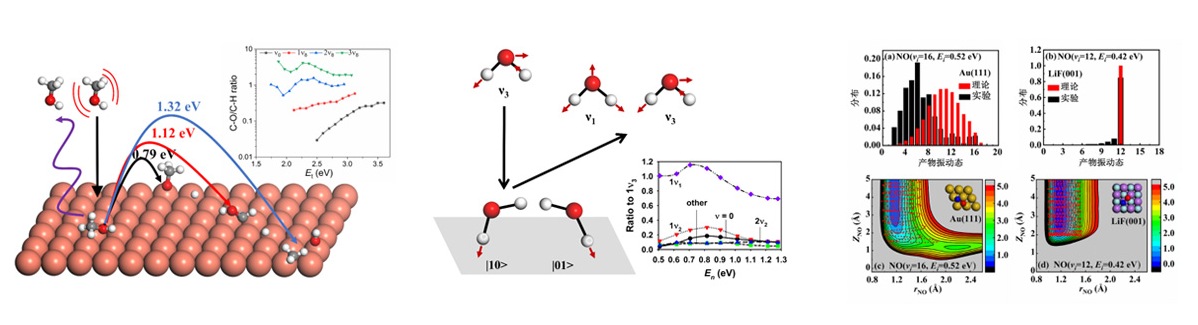
Chemical reactions on surfaces, especially the dissociative chemisorption of molecules on metal surfaces, are of great importance in heterogeneous catalysis. Based on global PESs, we perform both quantum and classical mechanical simulations to examine the reaction dynamics of these processes. We study how the initial rovibrational excitation or orientation of the molecule (or a particular bond) affects its reactivity on the surfaces, corresponding to very fundamental physicochemical problems like mode specificity, bond selectivity, and steric effects7. The influential factors of surface reactions also include the motion of surface atoms and the energy transfer between the molecule and surface, which need to be taken into account into our dynamical models. We develop quantum waveapacket methods to solve the time-dependent Schrodinger equation incorporating molecular degrees of freedom (DOFs), particularly for polyatomic molecules8. Nevertheless, the computational cost and the required memory of the quantum model scale exponentially with the number of DOFs. So we also develop much more efficient quasi-classical and semi-classical methods (e.g. Adiabatic switching)9 to include all molecular and surface DOFs. More recently, we start to use path-integral based ring polymer molecular dynamics method to study gas-surface reaction dynamics10. With these theoretical efforts, we try to unravel new mechanisms and dynamics and distill general rules of thumb from these complex systems. Recent examples include H2O scattering from Cu(111)11, CH3OH dissociative chemisorption on Cu(111)12, NO scattering from Au(111)13-15, and so on.
References:
7、B. Jiang, M. Yang, D. Xie, and H. Guo, Chem. Soc. Rev. 45, 3621 (2016).
8、B. Jiang, Chem. Sci. 8, 6662 (2017).
9、L. Zhang, J. Chen, and B. Jiang, J. Phys. Chem. C 125, 4995 (2021).
10、Q. Liu, L. Zhang, Y. Li, and B. Jiang, J. Phys. Chem. Lett. 10, 7475 (2019).
11、L. Zhang, and B. Jiang, Phys. Rev. Lett. 123, 106001 (2019).
12、J. Chen, X. Zhou, Y. Zhang, and B. Jiang, Nat. Commun. 9, 4039 (2018).
13、R. Yin, Y. Zhang, and B. Jiang, J. Phys. Chem. Lett. 10, 5969 (2019).
14、C. L. Box, Y. Zhang, R. Yin, B. Jiang, and R. J. Maurer, JACS Au 1, 164 (2021).
15、R. Yin, and B. Jiang, Phys. Rev. Lett. 126, 156101 (2021).