Phonon Dispersion of Rutile TiO$_2$
This is a summary of my using Phonopy to calculate phonon spectrum of rutile TiO$_2$. 1
Computational Details
Below is a list of used packages and computational details
-
DFT code: VASP version 5.4.4
-
PAW method, GGA (PBE).
Click here to download the POTCAR I used, where normalTiPOTCAR (4 valence electrons) was used.Note that if
Ti_svPOTCAR (12 valence electrons) is used instead, there will be three imaginary modes at Gamma point, one of which resembles the A2u mode that is responsible for the incipient ferroelectric behavior in rutile TiO$_2$. To remove the imaginary modes, eitherDFT+UorLDAshould be used.The lattice constants from different
POTCARand functional:-
PBE+TiPOTCAR:$a = 4.662\,\mathring{A};\thinspace c=2.969\,\mathring{A}$ and NO imaginary modes at Gamma point.
-
PBE+Ti_svPOTCAR:$a = 4.645\,\mathring{A};\thinspace c=2.967\,\mathring{A}$ and three imaginary moes at Gamma point
-
LDA+Ti_svPOTCAR:$a = 4.554\,\mathring{A};\thinspace c=2.922\,\mathring{A}$ and NO imaginary modes at Gamma point.
-
-
ASE for lattice constants optimization.
-
Matplotlib for plotting. 2
Step by Step Calculation
Optimizing the Lattice Constants
A script utilizing ASE to optimize the lattice constant is
used.3 As is shown below, the python script is embedded in the
job submission script.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
#!/bin/bash
#SBATCH --partition h3c
#SBATCH --time 12:00:00
#SBATCH --nodes 1
#SBATCH --ntasks-per-node 48
#SBATCH --cpus-per-task 1
#SBATCH --error out_%j.err
#SBATCH --output out_%j.log
ulimit -s unlimited
module load vasp/5.4.4
#module load vasp/5.4.4_soc
# module load vasp/6.1.0
#module load vasp/6.1.0_omp
echo "============================================================"
echo "Job ID: $SLURM_JOB_NAME"
echo "Job name: $SLURM_JOB_NAME"
echo "Number of nodes: $SLURM_JOB_NUM_NODES"
echo "Number of processors: $SLURM_NTASKS"
echo "Task is running on the following nodes:"
echo $SLURM_JOB_NODELIST
echo "OMP_NUM_THREADS = $SLURM_CPUS_PER_TASK"
echo "============================================================"
echo
python << EOF
import os
import numpy as np
from ase.io import read, write
from ase.calculators.vasp import Vasp
from ase.constraints import UnitCellFilter
from ase.optimize import BFGS as QuasiNewton
os.environ['VASP_COMMAND'] = "srun -n ${SLURM_NPROCS} vasp_std"
os.environ['VASP_PP_PATH'] = "/public/apps/vasp/pp/"
# the starting configuration
geo = read('init.vasp', format='vasp')
# setting the c-axis parameter
# geo.cell[-1,-1] = 25
calc = Vasp(
###############################
# General
###############################
system = 'vaspCalc',
prec = 'Accurate',
encut = 500,
ispin = 1,
istart = 0,
icharg = 1,
###############################
# Electronic relaxation
###############################
# isym = 0,
algo = 'Fast',
nelmin = 4,
nelm = 120,
xc = 'pbe',
ediff = 1E-8,
ismear = 0,
sigma = 0.04,
npar = 4,
###############################
# k-points grid
###############################
kpts = (15, 15, 23),
gamma = True,
###############################
# Writing Flat
###############################
nwrite = 1,
lwave = False,
lcharg = False,
###############################
# Vdw Correction
###############################
# ivdw = 10,
###############################
# dipole correction
###############################
# ldipol = True,
# idipol = 3,
# dipol = [0.5, 0.5, 0.5]
)
geo.set_calculator(calc)
# stress mask [XX, YY, ZZ, YZ, XZ, XY]
# 0 if fixed else 1
unf = UnitCellFilter(geo, mask=[1,1,1,1,1,1])
# unf = UnitCellFilter(geo, mask=[1,1,0,0,0,1])
dyn = QuasiNewton(unf, logfile='opt.log', trajectory='opt.traj')
dyn.run(fmax=0.001)
write('final.vasp', geo, vasp5=True, direct=True)
EOF
A few notes
-
The
INCAR,KPOINTSandPOTCARare automatically generated by the script based on the input settings. Therefore, there is NO NEED to prepare these files. -
The initial geometry is read from the file
init.vaspandPOSCARwill be automatically generated byASE. -
Note that the inputs parameters in the script are basically a
SCFcalculation, i.e.IBRION = -1. -
The stopping criteria is in the line
dyn.run(fmax=0.001), and the optimized geometry is written in the filefinal.vasp
Born Effective Charge and Phonon at Gamma-point
As TiO$_2$ is an ionic crystal, there are LO-TO splittings in the optical phonons. One must perform non-analytical (NAC) term correction by calculating Born effective charge.
-
I used DFPT to get the Born effective charge as well as the phonon at $\Gamma$-point.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27
General SYSTEM = VaspCalc PREC = Accurate ENCUT = 500 eV ISPIN = 1 ISTART = 1 ICHARG = 1 Ionic Relaxation IBRION = 8 Electronic relaxation ALGO = Fast NELMIN = 4 NELM = 100 EDIFF = 1E-8 ISMEAR = 0 SIGMA = 0.04 LORBIT = 11 LREAL = .FALSE. ADDGRID = .TRUE. Writing Flag NWRITE = 1 LWAVE = .FALSE. LCHARG = .FALSE. LEPSILON = .TRUE.
Note the last line is the extra setting needed to get Born effective charge.
-
When the calculation is finished, used
phonopy-vasp-bornto get the Born effective charge1 2 3 4 5 6
phonopy-vasp-born | tee BORN # epsilon and Z* of atoms 1 3 7.95986424 0.00000000 0.00000000 0.00000000 7.95986424 0.00000000 0.00000000 0.00000000 9.77308295 6.48351229 1.12137427 0.00000000 1.12137427 6.48351229 0.00000000 0.00000000 0.00000000 8.13997738 -3.24175614 1.91676778 0.00000000 1.91676778 -3.24175614 0.00000000 0.00000000 0.00000000 -4.06998869
-
One can see that there are no negative (imaginary) frequencies, except for three small negative ones which corresponds to translational motion, at the $\Gamma$-point. This means that we have get a fairly optimized cell.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
grep THz OUTCAR 1 f = 23.488597 THz 147.583210 2PiTHz 783.495252 cm-1 97.141066 meV 2 f = 17.390776 THz 109.269470 2PiTHz 580.093838 cm-1 71.922495 meV 3 f = 14.411095 THz 90.547583 2PiTHz 480.702388 cm-1 59.599522 meV 4 f = 14.411095 THz 90.547583 2PiTHz 480.702388 cm-1 59.599522 meV 5 f = 13.186496 THz 82.853200 2PiTHz 439.854157 cm-1 54.534985 meV 6 f = 13.186496 THz 82.853200 2PiTHz 439.854157 cm-1 54.534985 meV 7 f = 12.217729 THz 76.766257 2PiTHz 407.539569 cm-1 50.528485 meV 8 f = 10.984280 THz 69.016267 2PiTHz 366.396131 cm-1 45.427347 meV 9 f = 10.667164 THz 67.023770 2PiTHz 355.818290 cm-1 44.115861 meV 10 f = 10.667164 THz 67.023770 2PiTHz 355.818290 cm-1 44.115861 meV 11 f = 4.322348 THz 27.158111 2PiTHz 144.177992 cm-1 17.875799 meV 12 f = 2.690268 THz 16.903451 2PiTHz 89.737671 cm-1 11.126057 meV 13 f = 2.690268 THz 16.903451 2PiTHz 89.737671 cm-1 11.126057 meV 14 f = 2.592941 THz 16.291927 2PiTHz 86.491191 cm-1 10.723545 meV 15 f = 2.393622 THz 15.039568 2PiTHz 79.842622 cm-1 9.899227 meV 16 f/i= 0.005079 THz 0.031909 2PiTHz 0.169401 cm-1 0.021003 meV 17 f/i= 0.005079 THz 0.031909 2PiTHz 0.169401 cm-1 0.021003 meV 18 f/i= 0.005766 THz 0.036231 2PiTHz 0.192342 cm-1 0.023847 meV
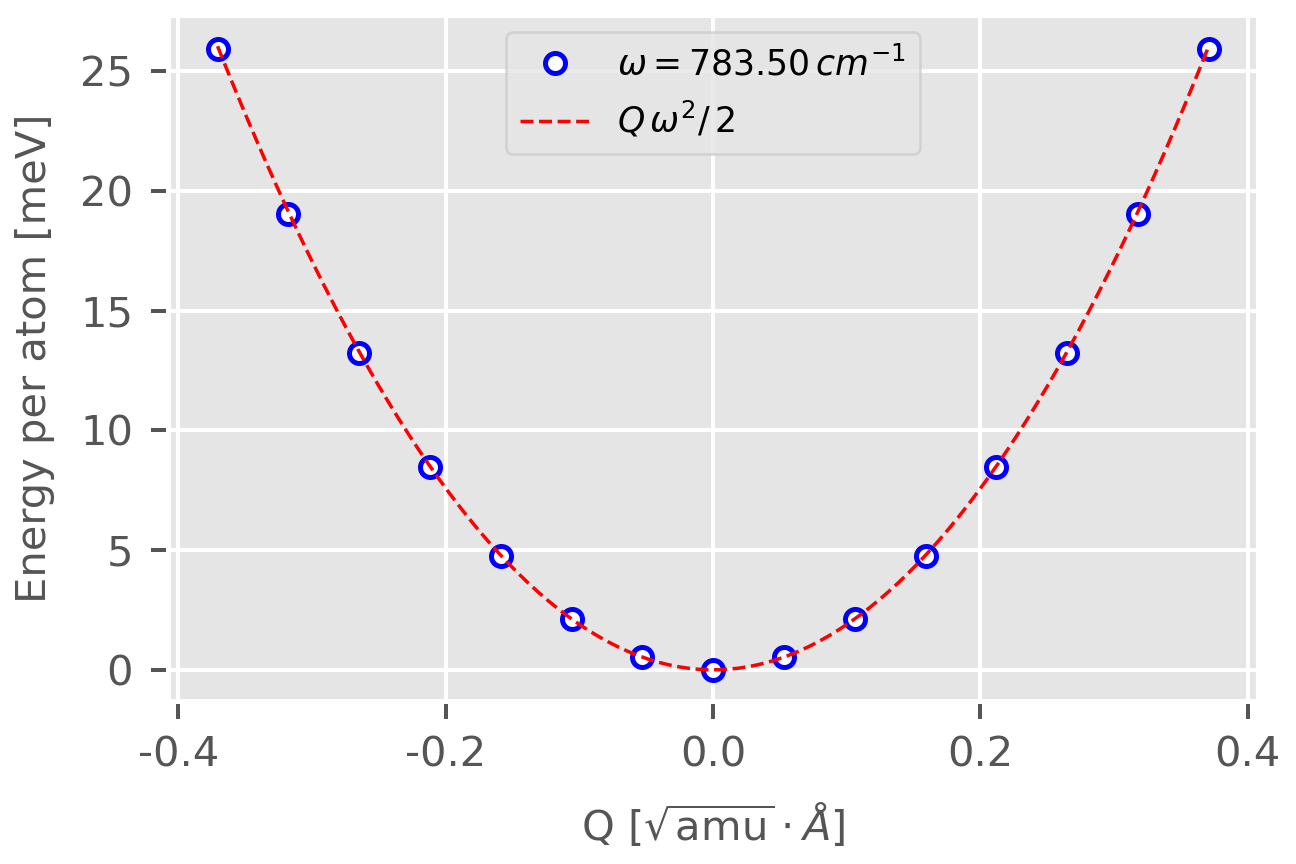
One can show that the Potential Energy Surface (PES) along the normal mode direction is indeed a harmonic potential.
-
Under the directory which you have performed the Gamma-point phonon calculation, use the script
init.shto initialize and generate a series of displacements along the normal mode (by the script phonon_traj.py).1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28
#!/bin/bash if [ -f OUTCAR.gz ]; then gzip -d OUTCAR.gz fi rm -f m_??/traj*.vasp 2>/dev/null for mode in {01..03} # select the modes do mkdir -p m_${mode} cd m_${mode} (( m = mode - 1 )) ./phonon_traj.py -i ../OUTCAR -p ../POSCAR -m ${m} -t 300 -msd classical --linear_traj -nsw 15 for ii in {01..15} do mkdir -p ${ii} cd ${ii} ln -sf ../../INCAR_scf INCAR ln -sf ../../POTCAR ln -sf ../../KPOINTS ln -sf ../traj_${ii}.vasp POSCAR cd ../ done cd .. done gzip OUTCAR
In then script
phonon_traj.py, we have set the phonon vibration amplitude according to a classical harmonic oscillator (-msd classical) with termperature300Kelvin (-t 300). -
Perform SCF calculation in each sub-directories and then plot the result using this script. Below is the result for the first normal mode, from which one can see that the calculated data fit well with the harmonic potential and the maximum PES of $25.8\,\mathrm{meV}$ corresponds well to $T=300\,K$.

Figure 1. Potential energy surface along the first normal mode. Blue dots are results from first-principles calculations and red dashed line is the harmonic potential. Figure plotted by Matplotlib. -
Phonon Spectrum from Frozen-phonon Method 4
-
Prepare
INCAR,KPOINTS, andPOSCAR_u, use the scriptinit.shto initialize the jobs.1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
#!/bin/bash rm POSCAR-* 2>/dev/null phonopy -d --dim="3 3 4" -c POSCAR_u for pos in POSCAR-* do mkdir -p d_${pos/POSCAR-} cd d_${pos/POSCAR-} ln -sf ../INCAR ln -sf ../POTCAR ln -sf ../KPOINTS cp ../${pos} POSCAR cd .. done
The script will create one directory for each displacement
POSCAR-00?. -
After the calculation in each displacement directory is done, prepare inputs for
phonopyto get the phonon band and DOS.Phonon band:
band.conf1 2 3
DIM = 3 3 4 BAND = AUTO EIGENVECTORS = .TRUE.
Phonon DOS:
dos.conf1 2 3 4 5
DIM = 3 3 4 MP = 51 51 51 DOS = .TRUE. GAMMA_CENTER = .TRUE. FPITCH = 0.02
DO NOT forget to copy the
BORNfile to current directory to perform NAC term correction. -
Use the script
pp.shto create theFORCE_SETSand calculation phonon band and DOS.1 2 3 4 5 6 7 8
#!/bin/bash if [ ! -f FORCE_SETS ]; then phonopy -f d_00?/vasprun.xml fi phonopy --nac band.conf phonopy --nac dos.conf python ph_band-dos.py # One can also use phonopy to plot the result.
In the script
pp.sh, apythonscriptph_band-dos.pyis used to plot the band and DOS in one figure, which of course can also be done byphonopy.The resulting output files:
One can use this site5 to visulize the animation of phonon modes by uploading the
band.yamlfile.
Phonon Band and Density-of-States
-
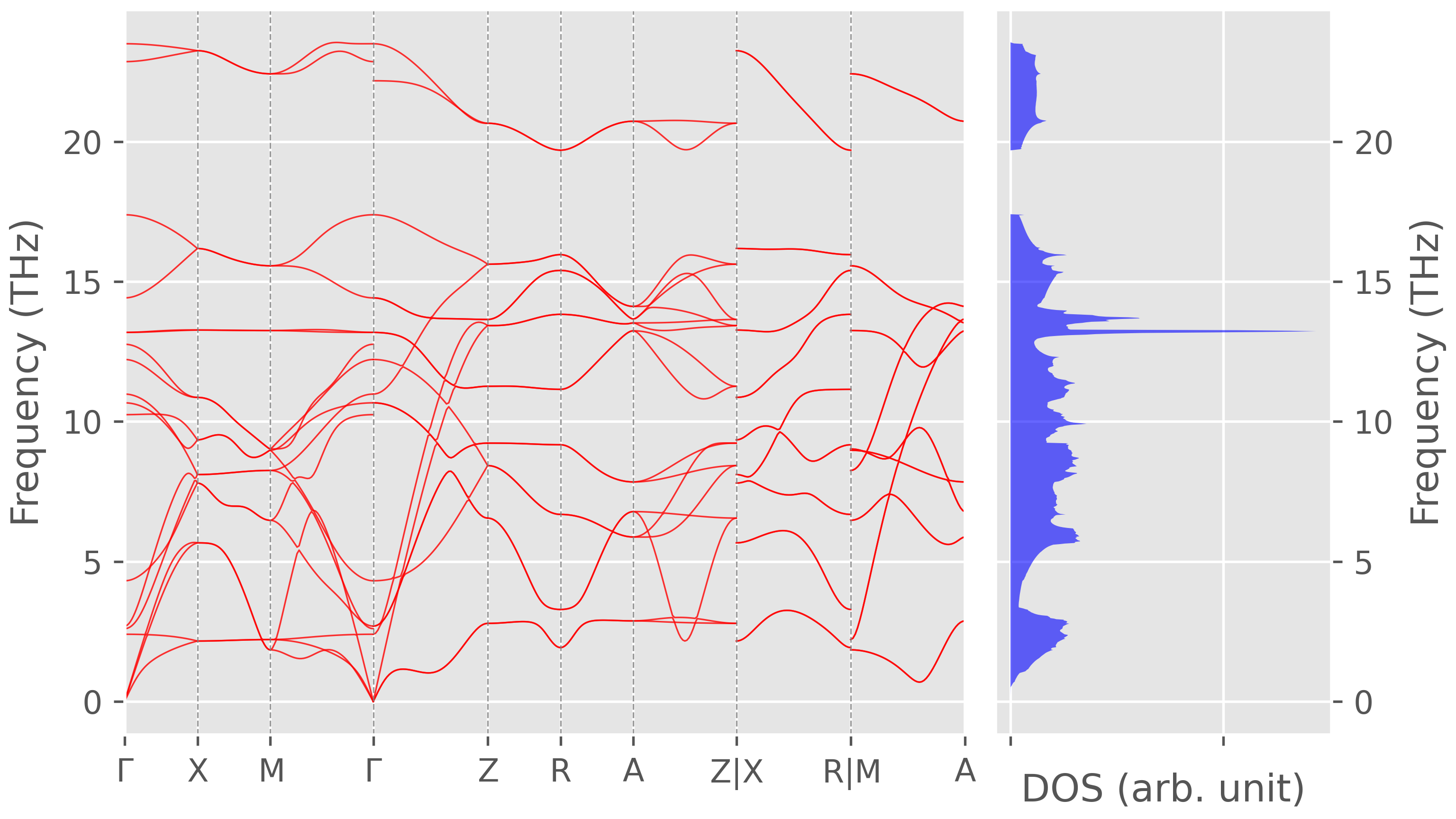
The phonon spectrum of rutile TiO$_2$ with NAC term correction
Figure 2. Phonon dispersion with non-analytical term correction. -
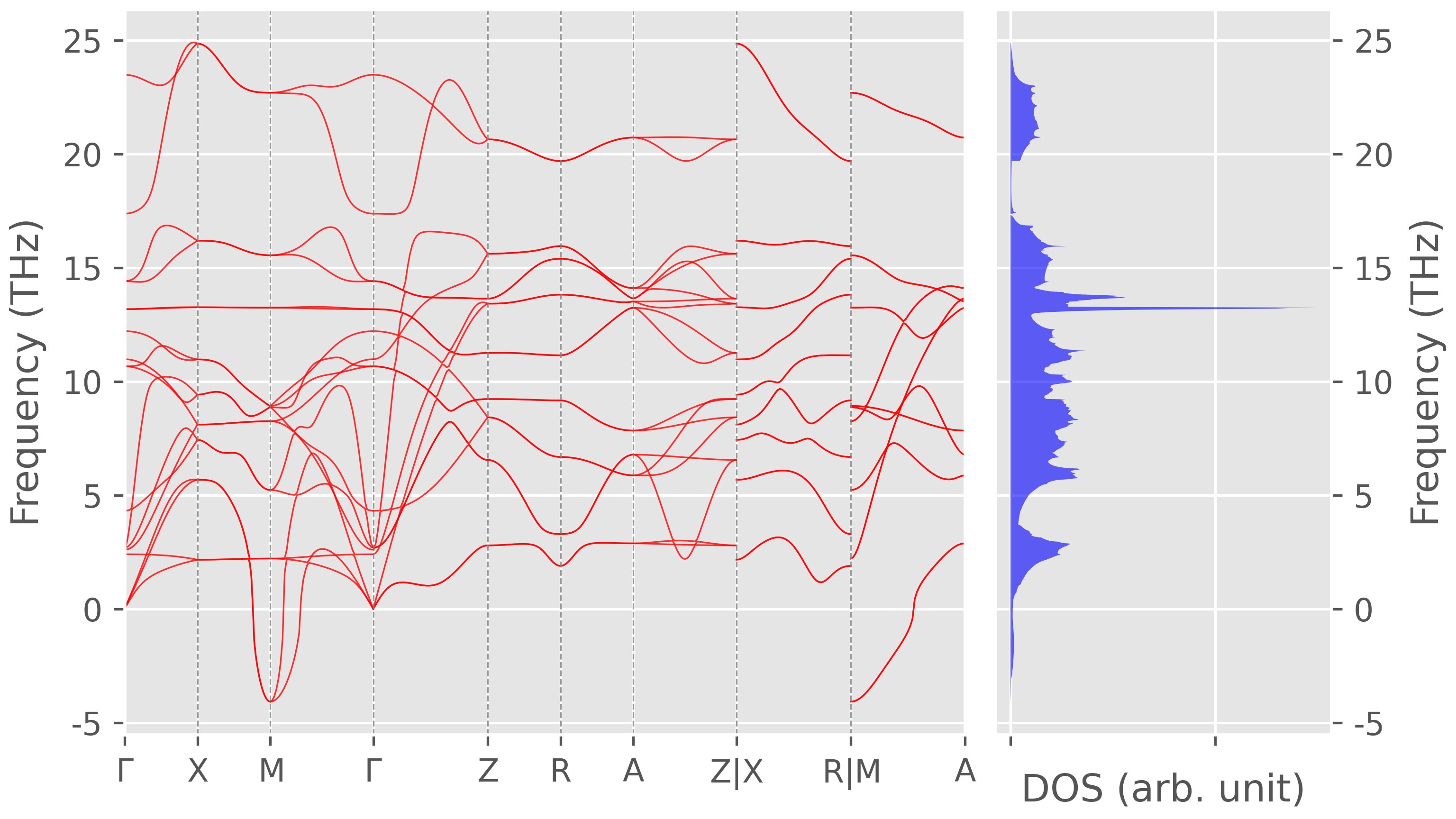
The phonon spectrum of rutile TiO$_2$ without NAC term correction, i.e. remove
--nacin thepp.sh.
Figure 3. Phonon dispersion without non-analytical term correction. -
One can see that the NAC term correction removes the imaginary frequencies along some $k$-path and generate LO-TO splitting at $\Gamma$ points.
-
Due to the NAC term, the phonon frequencies at $\Gamma$ point depend on the direction of $\mathbf{q} \to 0$. As a result, the result of Raman spectra depend on the angle between the incident light and the sample.
-
BTW, I thought NAC term correction only affects the phonons near $\Gamma$-point, which seems to be untrue,6 as
phonopyalso perform long-range dipole-dipole correction at general $q$-points.7
-
-
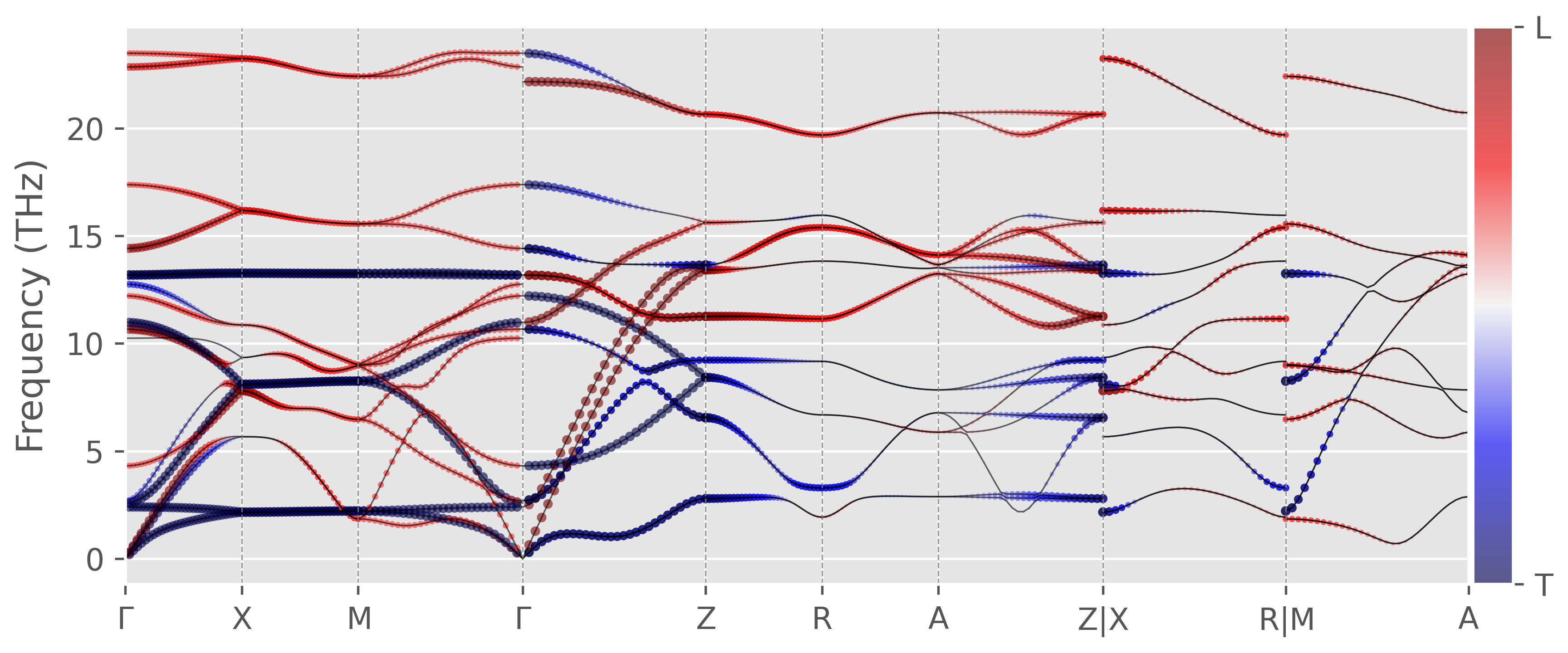
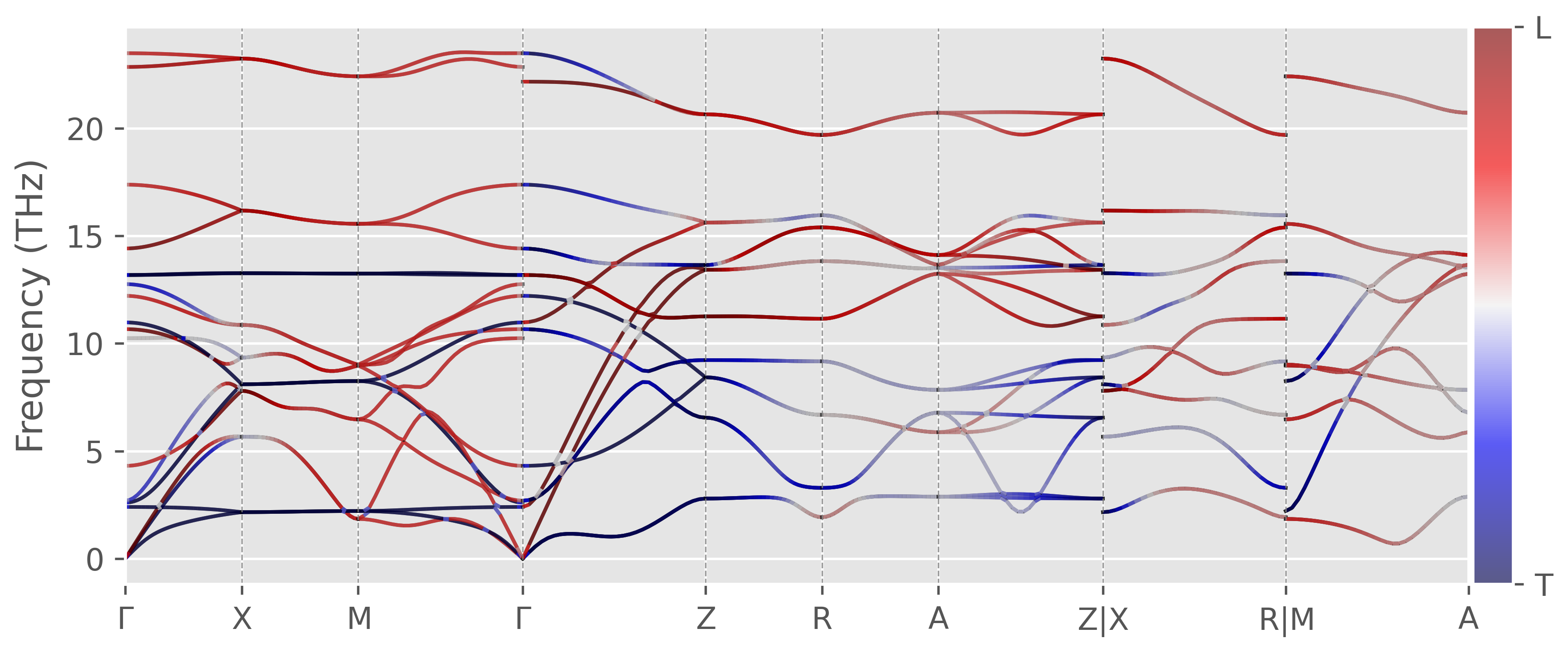
Longitudinal and Tranverse phonon modes
For purely longitudinal phonon mode, the polarization vector of the mode $\mathbf{p}_{nq}$ is parallel to the wavevector $\mathbf{q}$, i.e.
\[\begin{equation*} W_{nk} = \dfrac{\mathbf{p}_{nk}\cdot\mathbf{q}}{|\mathbf{q}|} = 1 \end{equation*}\]On the other hand, the polarization vector of a purely transverse mode is perpendicular to the wavevector, or
\[\begin{equation*} W_{nk} = \dfrac{\mathbf{p}_{nk}\cdot\mathbf{q}}{|\mathbf{q}|} = 0 \end{equation*}\]In practice, the classification of purely longitudinal and transverse modes is only valid for isotropic materials. For high symmetry structures (like cubic) and in some high symmetry directions, it is also a very good approximation. But for lower symmetry structures you cannot classify modes as purely transverse or longitudinal. Nevertheless, we can use $W_{nk}$ to quantify the similarness of the mode to longitudinal (close to 1) or transverse modes (close to 0).
Figure 4. Rutile TiO2 phonon dispersion overlapped with Wnk, generated by this script. The same result with color-stripe plot
Figure 5. Rutile TiO2 phonon dispersion overlapped with Wnk, generated by this script.